Secondary metabolite discovery
Under Development!
This tutorial is not in its final state. The content may change a lot in the next months. Because of this status, it is also not listed in the topic pages.
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How to discover secondary metabolites produced by microorganisms ?
How to identify the discovered secondary metabolites in compound libraries ?
How to bridge workflow steps using custom scripts ?
Requirements:
Gene cluster prediction using antiSMAHS.
Extraction of gene cluster products using a custom script.
Query of the products in compound libraries using cheminformatic tools.
Time estimation: 3 hoursSupporting Materials:Published: Feb 21, 2024Last modification: Sep 27, 2024License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00420version Revision: 2
Genome mining is an important source for various bio-active compounds such as antibiotics and fungicides (Adamek et al. 2017). One challenge in secondary metabolite discovery using genome mining requires to annotate biosynthetic gene clusters (BGCs) using dedicated tools such as antiSMASH (Blin et al. 2023) as well as to query the discovered secondary metabolites against compound libraries (Zierep et al. 2020) in order to identify weather they might posses bio-active properties.
In this toturial we will:
- Annotate biosynthetic gene clusters (BGCs) using antiSMASH.
- Extract the predicted compounds as
SMILESusing a custom script. - Query the predicted compounds agains the MIBiG (Terlouw et al. 2023) which is a dedicated BGC database.
- Characterize the predicted compounds using dedicated cheminformatic tools.
AgendaIn this tutorial, we will cover:
- Galaxy and data preparation
- Biosynthetic gene clusters (BGCs) detection and compound extraction
- Use custom scripts in Galaxy to bridge between workflow steps
- Cheminformatics
- Convert the table into a two column table
- Remove duplicated molecules
- Generate fingerprints for the predicted secondary metabolites and the target database with Molecule to fingerprint
- Compare prediction with target DB using Similarity Search
- Determine the novelty of the compounds using Natural Product likeness calculator
- Check Drug-likeness of the predicted compounds
- Conclusion
Galaxy and data preparation
Any analysis should get its own Galaxy history. So let’s start by creating a new one and get the data into it.
Get data
Hands On: History Creation and Data Upload
Create a new history for this tutorial
To create a new history simply click the new-history icon at the top of the history panel:
Rename the history
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name
- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name
- Press Enter
Import the files from Zenodo or from the shared data library (
GTN - Material->genome-annotation->Secondary metabolite discovery):https://zenodo.org/record/10652998/files/MIBiG_compounds_3.0.sdf https://zenodo.org/record/10652998/files/gbk2features.ipynbFor this training we need a custom jupyter notebook and a compound library.
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
- Rename the datasets
Check that the datatype is correct
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
datatypesfrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
Add useful tags to your datasets
Datasets can be tagged. This simplifies the tracking of datasets across the Galaxy interface. Tags can contain any combination of letters or numbers but cannot contain spaces.
To tag a dataset:
- Click on the dataset to expand it
- Click on Add Tags galaxy-tags
- Add tag text. Tags starting with
#will be automatically propagated to the outputs of tools using this dataset (see below).- Press Enter
- Check that the tag appears below the dataset name
Tags beginning with
#are special!They are called Name tags. The unique feature of these tags is that they propagate: if a dataset is labelled with a name tag, all derivatives (children) of this dataset will automatically inherit this tag (see below). The figure below explains why this is so useful. Consider the following analysis (numbers in parenthesis correspond to dataset numbers in the figure below):
- a set of forward and reverse reads (datasets 1 and 2) is mapped against a reference using Bowtie2 generating dataset 3;
- dataset 3 is used to calculate read coverage using BedTools Genome Coverage separately for
+and-strands. This generates two datasets (4 and 5 for plus and minus, respectively);- datasets 4 and 5 are used as inputs to Macs2 broadCall datasets generating datasets 6 and 8;
- datasets 6 and 8 are intersected with coordinates of genes (dataset 9) using BedTools Intersect generating datasets 10 and 11.
Now consider that this analysis is done without name tags. This is shown on the left side of the figure. It is hard to trace which datasets contain “plus” data versus “minus” data. For example, does dataset 10 contain “plus” data or “minus” data? Probably “minus” but are you sure? In the case of a small history like the one shown here, it is possible to trace this manually but as the size of a history grows it will become very challenging.
The right side of the figure shows exactly the same analysis, but using name tags. When the analysis was conducted datasets 4 and 5 were tagged with
#plusand#minus, respectively. When they were used as inputs to Macs2 resulting datasets 6 and 8 automatically inherited them and so on… As a result it is straightforward to trace both branches (plus and minus) of this analysis.More information is in a dedicated #nametag tutorial.



Biosynthetic gene clusters (BGCs) detection and compound extraction
The general idea of this tutorial is described in the flowchart below. Zierep et. al. previously created a workflow that combines BGC prediction with natural compound screening in their webserver SeMPI 2.0 (Zierep et al. 2020). Here, we want to reproduce the workflow using Galaxy, which allows to adjust the workflow and combine it with other tools and databases. E.g. the workflow could be combined with metagenomic workflows, that allow to screen a vast amount of metagenome assembly genomes (MAGs) for potential novel bioactive compounds.
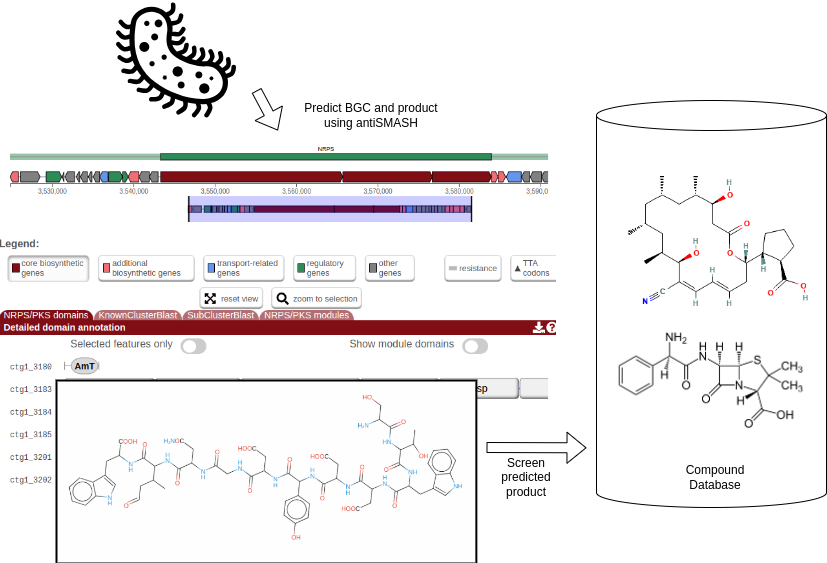 Open image in new tab
Open image in new tabDownload a bacterial genome with NCBI Accession Download
Hands On: Task description
- NCBI Accession Download ( Galaxy version 0.2.8+galaxy0) with the following parameters:
- “Select source for IDs”:
Direct Entry
- “ID List”:
AL645882.2- “Molecule Type”:
NucleotideComment: Genome downloadThis downloads the
Streptomyces coelicolor A3(2) complete genome, which should be a great source for biosynthetic gene clusters (BGCs).
Detect BGCs and predict NRPS / PKS metabolite structures with Antismash
Hands On: Task description
- Antismash ( Galaxy version 6.1.1+galaxy1) with the following parameters:
- param-file “Sequence file in GenBank,EMBL or FASTA format”:
Downloaded Files(output of NCBI Accession Download tool)- “Taxonomic classification of input sequence”:
BacteriaComment: BGC detectionThis step uses Antismash for BGC detection. Another tool that could be used is prism. But prism can only be used via a web server and is therefore not integrable into Galaxy workflows.
Question
- How many BGCs are detected for the
Streptomyces coelicolor A3(2) complete genome?- How many BGCs are of type NRPS ?
- In the HTML report of Antismash you can find 27 BGCs of various types.
- Of those 27 BGCs 4 are pure NRPS and one more is classified as NRPS-like.
Collapse single BGC Genbank files into one with Collapse Collection
Hands On: Task description
- Collapse Collection ( Galaxy version 5.1.0) with the following parameters:
- param-file “Collection of files to collapse into single dataset”:
Genbank(output of Antismash tool)- “Prepend File name”:
YesComment: Collapse Genbank filesAntismash produces one Genbank file for each cluster. Genbank files can contain multiple records. For downstream analysis it is more convenient to combine all clusters into one file.
Use custom scripts in Galaxy to bridge between workflow steps
Integrate a custom script into the workflow using Interactive JupyTool and notebook
For the next steps we need to extract the SMILES representations of the predicted BGC metabolites.
And convert it into the .smi format (basically a tabular format that contains names and structures of chemical compounds).
The SMILES representations of the predicted BGC metabolites are stored as features of the clusters in the Genbank files.
cand_cluster 1..49828
/SMILES="NC(CCCN)C(=O)NC(C(O)C)C(=O)NC(CCCN)C(=O)O"
/candidate_cluster_number="1"
/contig_edge="False"
/detection_rules="cds(Condensation and (AMP-binding or
A-OX))"
/kind="single"
/product="NRPS"
/protoclusters="1"
/tool="antismash"
There are various tools that can convert Genbank files into GFF (like customGbkToGff ( Galaxy version 20.1.0.0) and genbank2gff3 ( Galaxy version 1.1)). From the GFF one could extract the SMILES string via regex. However, unfortunately both tools seem to have difficulty to parse SMILES and introduce artefacts like:
SMILES=CC(%3DO)C(%3DO)O
Therefore we need to quickly prototype our own script, that allows to generate the desired SMILES representations.
This is in fact a great chance to demonstrate how the interactive Interactive JupyTool and notebook can be used to connect workflow steps, for which there is no dedicated tool yet available in Galaxy, showing the flexible nature of Galaxy.
Hands On: Task description
- Interactive JupyTool and notebook with the following parameters:
- “Do you already have a notebook?”:
Load a previous notebook
- param-file “IPython Notebook”:
gbk2features.ipynb(from your history)- “Execute notebook and return a new one.”:
Yes- In “User inputs”:
- param-repeat “Insert User inputs”
- “Name for parameter”:
dataset(a file calleddatasetwill be created in the notebook in the foldergalaxy_inputs)- “Choose the input type”:
Dataset
- param-file “Select value”:
Collapse Genbank files(output of Collapse Collection tool)Comment: Interactive JupyTool and notebookJupyTool notebooks can be executed interactively, which allows to run any code (python / R / bash) dynamically or as a normal tool itself, that executes a provide script and return the output. More information about the JupyTool can be found in the Interactive JupyTool Tutorial.
In this workflow the jupyter notebook is executed and the output collected. If you want to run the notebook interactively. Rerun the tool and set the option “Execute notebook and return a new one.”: No. Now the notebook will start up interactively and you can play around the the python code.
The code we provide for the notebook looks like this:
from Bio import SeqIO
import pandas as pd
import numpy as np
input_path = "galaxy_inputs"
use_qualifiers = ["SMILES"]
feature_list = []
for folder in os.listdir(input_path):
folder_path = os.path.join(input_path, folder)
if os.path.isdir(folder_path):
for file in os.listdir(folder_path):
file_path = os.path.join(folder_path, file)
if '.genbank' in file:
for record in SeqIO.parse(file_path, "genbank"):
#print(record)
for feat in record.features:
combined_features = {}
combined_features['ID'] = record.id
combined_features['Name'] = record.name
combined_features['Type'] = feat.type
combined_features['Start'] = feat.location.start
combined_features['End'] = feat.location.end
combined_features['Strand'] = feat.location.strand
for qualifier in feat.qualifiers.items():
combined_features[qualifier[0]] = qualifier[1][0]
feature_list.append(combined_features)
df = pd.DataFrame(feature_list)
# df trimming is simpler with pandas
df.replace('', np.nan, inplace=True)
df = df.dropna(subset=use_qualifiers)
#remove extra space antiSMASH bug (https://github.com/antismash/antismash/issues/694)
df['SMILES'] = df['SMILES'].str.replace(' ', '')
#only keep what is needed downstream
df = df.loc[:,['ID', 'Name', 'Type', 'Start', 'End', 'Strand', 'SMILES']]
df.to_csv("outputs/collection/feature_table.tsv", sep="\t")
Question
- What does the notebook do ?
- The notebook creates a table with a row for each
SMILESqualifier.
Cheminformatics
Now, that we have predicted the products of the BGCs found in the Streptomyces coelicolor A3(2) complete genome,
we can compare the chemical structures of the prediction with natural compound libraries to see if some of the compounds
are similar to other compounds with known bio-activte properties. Those compounds should be further investigated.
The natural compound library we are using here comprises all the compounds found in the MIBiG (Terlouw et al. 2023).
Convert the table into a two column table
The next two steps convert the generated table into the .smi format. The .smi format is basically a specific table format where the first column is a chemical structure and the second column the ID or name of the compound. The table must
not have a header.
Hands On: Table to smi format conversion
- Text reformatting ( Galaxy version 1.1.2) with the following parameters:
- param-file “File to process”:
output_collection(output of Interactive JupyTool and notebook tool)- “AWK Program”:
{print $8, $2-$5-$6}- Remove beginning with the following parameters:
- param-file “from”:
outfile(output of Text reformatting tool)Now, that the table is indeed the same as the
.smiformat it can also be converted into.smi.
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
smifrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button
Remove duplicated molecules
Hands On: Remove duplicated molecules
- Remove duplicated molecules ( Galaxy version 3.1.1+galaxy0) with the following parameters:
- param-file “Molecular input file”: (output of Remove beginning tool)
- “Select descriptor for molecule comparison”:
Canonical SMILESComment: short descriptionFor the downstream analysis deduplicated molecules are not required. Especially short, hybrid or uncompleted cluster can produce short molecular structures, that appear multiple times in an organisms. These structures can be regarded as artefacts and could probably be removed as well.
Generate fingerprints for the predicted secondary metabolites and the target database with Molecule to fingerprint
Various molecular fingerprint options can be used to convert the compounds. Details about molecular fingerprint can be found in Muegge and Mukherjee 2016 and Capecchi et al. 2020.
Hands On: Task description
- Molecule to fingerprint ( Galaxy version 1.5) with the following parameters:
- param-file “Molecule file”: (output of Remove duplicated molecules tool)
- “Type of fingerprint”:
Open Babel FP2 fingerprints- Molecule to fingerprint ( Galaxy version 1.5) with the following parameters:
- param-file “Molecule file”:
output(MIBiG_compounds_3.0.sdf)- “Type of fingerprint”:
Open Babel FP2 fingerprintsComment: Be consitentA major application of molecular fingerprints is to determine the similarity of compounds. For the downstream tool Similarity Search ( Galaxy version 0.2) it is important that the same fingerprint are used for the query and target compounds!
Now that we have converted our compounds into molecular fingerprints, we can compare them to find the closest match between our prediction and the target database.
Compare prediction with target DB using Similarity Search
Hands On: Task description
- Similarity Search ( Galaxy version 0.2) with the following parameters:
- “Subject database/sequences”:
Chemfp fingerprint file
- “Query Mode”:
Query molecules are stores in a separate file
- param-file “Query molecules”: (output of Molecule to fingerprint for
MIBiG_compounds_3.0.sdftool)- param-file “Target molecules”: (output of Molecule to fingerprint from Remove duplicated molecules tool)
- “select the k nearest neighbors”:
1
Determine the novelty of the compounds using Natural Product likeness calculator
Normally, this tool allows to estimate how similar any kind of compound is to a natural product. In our case we know that the compounds are natural compounds. However, by comparing to a large source of known natural products we can estimate the novelty of our predicted structure. This could for example be used to mine metagenomic datasets for likely sources of uncharacterized molecular scaffolds.
Hands On: Task description
- Natural Product ( Galaxy version 2.1) with the following parameters:
- param-file “Molecule file”: (output of Remove duplicated molecules tool)
Check Drug-likeness of the predicted compounds
Estimates the drug-likeness of molecules, based on eight commonly used molecular properties, and reports a score between 0 (all properties unfavourable) to 1 (all properties favourable). Two possible methods to weight the features are available (QEDw,mo, QEDw,max), as well as an option to leave features unweighted (QEDw,u).
The eight properties used are: molecular weight (MW), octanol–water partition coefficient (ALOGP), number of hydrogen bond donors (HBDs), number of hydrogen bond acceptors (HBAs), molecular polar surface area (PSA), number of rotatable bonds (ROTBs), number of aromatic rings (AROMs) and number of structural alerts (ALERTS).
Hands On: Task description
- Drug-likeness ( Galaxy version 2021.03.4+galaxy0) with the following parameters:
- param-file “Molecule data in SDF or SMILES format”: (output of Remove duplicated molecules tool)
Conclusion
In this simple use case the combination of BGC prediction software and cheminformatics was used to detect and characterize novel secondary metabolites.
You've Finished the Tutorial
Key points
Gene cluster prediction that can be performed for any assembly or genome of microorganisms (procaryotes and fungi).
Bridging of gene annotation and cheminformatics.
Usage of custom script to bridge workflow steps.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsReferences
- Muegge, I., and P. Mukherjee, 2016 An overview of molecular fingerprint similarity search in virtual screening. Expert Opinion on Drug Discovery 11: 137–148. 10.1517/17460441.2016.1117070
- Adamek, M., M. Spohn, E. Stegmann, and N. Ziemert, 2017 Mining Bacterial Genomes for Secondary Metabolite Gene Clusters. Methods in Molecular Biology (Clifton, N.J.) 1520: 23–47. 10.1007/978-1-4939-6634-9_2
- Capecchi, A., D. Probst, and J.-L. Reymond, 2020 One molecular fingerprint to rule them all: drugs, biomolecules, and the metabolome. Journal of Cheminformatics 12: 43. 10.1186/s13321-020-00445-4
- Zierep, P. F., A. T. Ceci, I. Dobrusin, S. C. Rockwell-Kollmann, and S. Günther, 2020 SeMPI 2.0-A Web Server for PKS and NRPS Predictions Combined with Metabolite Screening in Natural Product Databases. Metabolites 11: 13. 10.3390/metabo11010013
- Blin, K., S. Shaw, H. E. Augustijn, Z. L. Reitz, F. Biermann et al., 2023 antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Research 51: W46–W50. 10.1093/nar/gkad344
- Terlouw, B. R., K. Blin, J. C. Navarro-Muñoz, N. E. Avalon, M. G. Chevrette et al., 2023 MIBiG 3.0: a community-driven effort to annotate experimentally validated biosynthetic gene clusters. Nucleic Acids Research 51: D603–D610. 10.1093/nar/gkac1049
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Paul Zierep, Secondary metabolite discovery (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/secondary-metabolite-discovery/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{genome-annotation-secondary-metabolite-discovery, author = "Paul Zierep", title = "Secondary metabolite discovery (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/secondary-metabolite-discovery/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/genome-annotation/tutorials/secondary-metabolite-discovery/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: antismash owner: bgruening revisions: bc88856eddab tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: chemfp owner: bgruening revisions: 73de2fe4bf92 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: natural_product_likeness owner: bgruening revisions: 6052dd3fa475 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: openbabel_remduplicates owner: bgruening revisions: c5de6c19eb06 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: simsearch owner: bgruening revisions: '0892f7ced10c' tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: cpt_gbk_to_gff owner: cpt revisions: a921d6148d88 tool_panel_section_label: Convert Formats tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bp_genbank2gff3 owner: iuc revisions: 11c3eb512633 tool_panel_section_label: Annotation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: ncbi_acc_download owner: iuc revisions: e063168e0a81 tool_panel_section_label: Get Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: collapse_collections owner: nml revisions: 90981f86000f tool_panel_section_label: Collection Operations tool_shed_url: https://toolshed.g2.bx.psu.edu/