Analysis of molecular dynamics simulations
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
Which analysis tools are available?
Requirements:
Learn which analysis tools are available.
Analyse a protein and discuss the meaning behind each analysis.
- Introduction to Galaxy Analyses
- tutorial Hands-on: Setting up molecular systems
- tutorial Hands-on: Running molecular dynamics simulations using NAMD
Time estimation: 1 hourLevel: Intermediate IntermediateSupporting Materials:Published: Jun 3, 2019Last modification: Nov 9, 2023License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00047rating Rating: 3.8 (0 recent ratings, 6 all time)version Revision: 13
Molecular dynamics simulations return highly complex data. The Cartesian positions of each atom of the system (thousands or even millions) are recorded at every time step of the trajectory; this may again be thousands to millions of steps in length. Therefore, some kind of further analysis is needed to extract useful information from the data.
In this tutorial, we illustrate some of the analytical tools able to investigate conformational changes by analysis of a typical short protein simulation, such as for CBH1.
There are other analysis tools available; you are encouraged to try these out too.
AgendaIn this tutorial, we will cover:
Get data
The data required can be generated by completing the NAMD simulation tutorial. Access it from your history. Alternatively, download the data from the Zenodo link provided.
Hands On: Upload cellulose simulation trajectory
Create a new history
To create a new history simply click the new-history icon at the top of the history panel:
Import the files from Zenodo, or from your history, if you completed the previous NAMD simulation tutorial:
https://zenodo.org/record/2537734/files/cbh1test.dcd https://zenodo.org/record/2537734/files/cbh1test.pdb
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
Rename the dcd file ‘CBH1 trajectory’ and rename the pdb file ‘CBH1 structure’
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, change the Name field
- Click the Save button


Analysis with BIO3D
We’ll carry out some basic analysis by calculating RMSD, RMSF and PCA. The tools use the Bio3D package, developed by the Grant lab.
RMSD
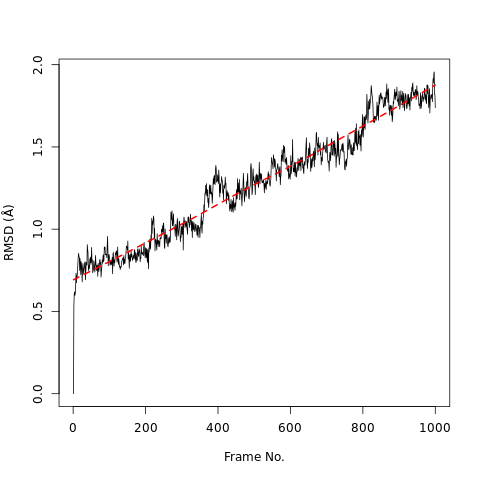
RMSD, or root-mean-square deviation, is a standard measure of structural distance between coordinates. It measures the average distance between a group of atoms (e.g. backbone atoms of a protein). If we calculate RMSD between two sets of atomic coordinates - for example, two time points from the trajectory - the value is a measure of how much the protein conformation has changed. Wikipedia provides more information.
Hands On: Calculate RMSDRMSD Analysis ( Galaxy version 2.3.4) with the following parameters:
- param-file “dcd trajectory input”: Trajectory file
- param-file “pdb input”: Structure file
- “Select domains”:
Calpha(calculate RMSD only for the C-alpha domain of the protein)
 Open image in new tab
Open image in new tab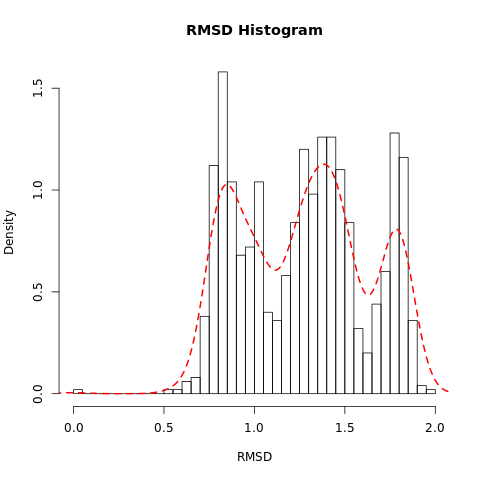 Open image in new tab
Open image in new tabQuestionWhat do the features in the RMSD plot tell us?
The increase in the RMSD plot with time shows the protein steadily deviates from its original conformation.
The three peaks visible in the histogram suggests the presence of three main conformations which are accessed during the trajectory.
RMSF
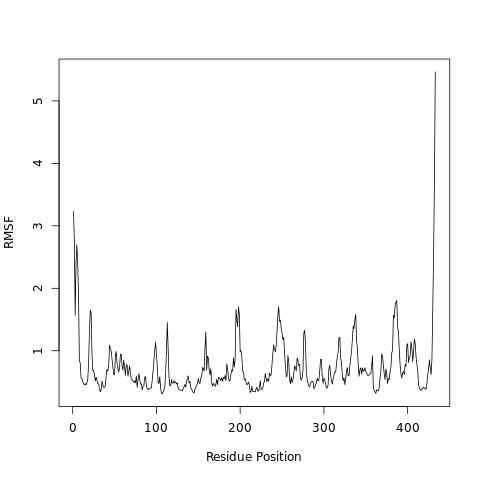
The root-mean-square fluctuation (RMSF) measures the average deviation of a particle (e.g. a protein residue) over time from a reference position (typically the time-averaged position of the particle). Thus, RMSF analyzes the portions of structure that are fluctuating from their mean structure the most (or least).
Hands On: Calculate RMSF
- RMSF Analysis ( Galaxy version 2.3.4) with the following parameters:
- param-file “dcd trajectory input”: Trajectory file
- param-file “pdb input”: Structure file
- “Select domains”:
Calpha(calculate RMSF only for the C-alpha domain of the protein)
 Open image in new tab
Open image in new tabQuestionWhat can we learn from the features in the RMSF plot?
Higher RMSF values most likely are loop regions with more conformational flexibility, where the structure is not as well defined.
This allows a link with experimental spectroscopic techniques which detect the secondary structure of a protein.
PCA
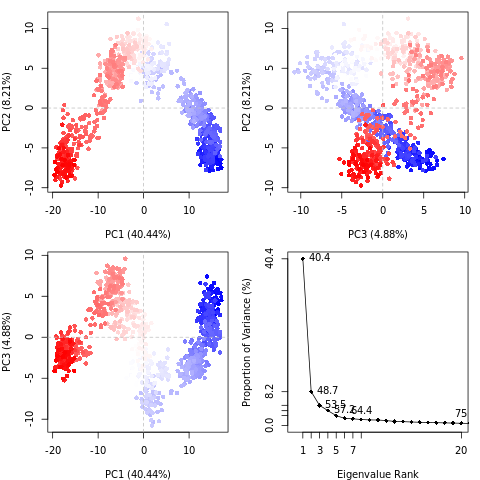
Principal component analysis (PCA) converts a set of correlated observations (movement of all atoms in protein) to a set of principal components which are linearly independent (or uncorrelated). Mathematically, it is a transformation of the data to a new coordinate system, in which the first coordinate represents the greatest variance, the second coordinate represents the second most variance, and so on.
You can read more about PCA on Wikipedia. In a nutshell, PCA takes a complex dataset with many variables and tries to distill the variables down to a few ‘principal components’ which still preserve most of the differences between the data.
In summary:
- The PCA tool tool will calculate and return a PCA to determine the relationship between statistically meaningful conformations (major global motions) sampled during the trajectory. THe tool returns several images of the PCA and the raw data in tab-separated format.
- The PCA visualization tool tool will carry out PCA and return a trajectory of the selected principle component. This trajectory is useful for visualisation and further investigating the interesting modes and changes that occur within a selected principle component.
Hands On: Calculate PCA
- PCA ( Galaxy version 2.3.4) with the following parameters:
- param-file “dcd trajectory input”: Trajectory file
- param-file “pdb input”: Structure file
- “Use singular value decomposition (SVD) instead of default eigenvalue decomposition ?”:
No- “Select domains”:
Calpha- PCA visualization ( Galaxy version 2.3.4) with the following parameters:
- param-file “dcd trajectory input”: Trajectory file
- param-file “pdb input”: Structure file
- “Use singular value decomposition (SVD) instead of default eigenvalue decomposition ?”:
No- “Select domains”:
Calpha- “Principal component id”:
1PCA visualisation: This tool can generate small trajectories of the first three principal components. The .pdb of the .nc files can be visualized using a visualization software such as VMD.
 Open image in new tab
Open image in new tabQuestionWhat do the features in the RMSD plot tell us? Do the principal coordinates have a meaning?
Here, PCA shows the statistically meaningful conformations in the CBH1 trajectory. The principal motions within the trajectory and the vital motions needed for conformational changes can be identified. Two distinct groupings along the PC1 plane, indicating a non-periodic conformational change, are identified. The groupings along the PC2 and PC3 planes do not completely cluster separately, implying that these global motions are periodic. The PC1 is linked to an active site motion that limits the motion to a key glycosidic bond.
Workflow vs. individual tools
You can choose to use the tools one by one as described above, or alternatively combine into a single analysis using the workflow provided.
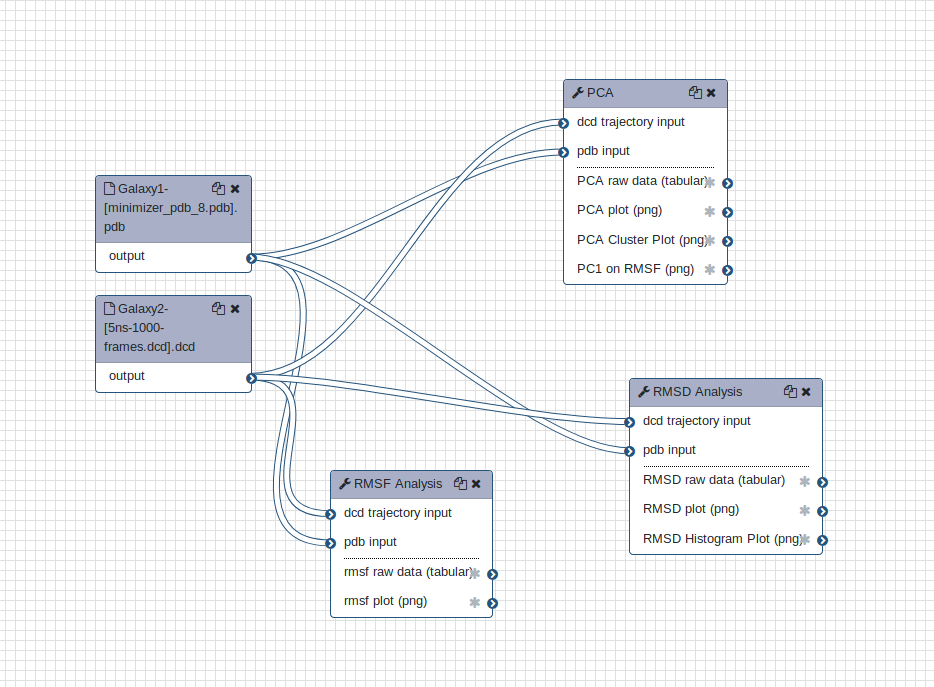 Open image in new tab
Open image in new tabHands On: Upload a workflow
Click on ‘Workflow’ in the toolbar at the top of the main Galaxy page. In the upper right corner of the central pane, click the ‘Upload or import workflow’ icon.
Enter the ‘Archived workflow URL’ and click ‘Import workflow’.
https://raw.githubusercontent.com/galaxyproject/training-material/master/topics/computational-chemistry/tutorials/analysis-md-simulations/workflows/main_workflow.ga
Further analysis
Further analyses are available; try out the MDAnalysis workflow, which includes a Ramachandran plot and various timeseries.
 Open image in new tab
Open image in new tabConclusion
You've Finished the Tutorial
Key points
Multiple analyses including timeseries, RMSD, PCA are available
Analysis tools allow a further chemical understanding of the system
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Christopher Barnett, Tharindu Senapathi, Simon Bray, Nadia Goué, Analysis of molecular dynamics simulations (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/analysis-md-simulations/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{computational-chemistry-analysis-md-simulations, author = "Christopher Barnett and Tharindu Senapathi and Simon Bray and Nadia Goué", title = "Analysis of molecular dynamics simulations (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/analysis-md-simulations/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Do you want to extend your knowledge?Follow one of our recommended follow-up trainings:
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/computational-chemistry/tutorials/analysis-md-simulations/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: bio3d_pca owner: chemteam revisions: 994c509873db tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bio3d_pca owner: chemteam revisions: 24867ab16f36 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bio3d_rmsd owner: chemteam revisions: f871c9a8cb7c tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bio3d_rmsd owner: chemteam revisions: 77e28e1da9f4 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bio3d_rmsf owner: chemteam revisions: 4354d92dc1aa tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bio3d_rmsf owner: chemteam revisions: 6bcb804a54c3 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: mdanalysis_cosine_analysis owner: chemteam revisions: ebeb72aa39a8 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: mdanalysis_dihedral owner: chemteam revisions: 554f60da9c8f tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: mdanalysis_distance owner: chemteam revisions: 489b25966bb9 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: mdanalysis_ramachandran_plot owner: chemteam revisions: d710c7f00ae6 tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: mdanalysis_rdf owner: chemteam revisions: 49dac57d004a tool_panel_section_label: ChemicalToolBox tool_shed_url: https://toolshed.g2.bx.psu.edu/