Durante la secuenciación, el secuenciador determina las bases nucleotídicas de una muestra de ADN o ARN (biblioteca). Para cada fragmento de la biblioteca se genera una secuencia, también llamada lectura, que no es más que una sucesión de nucleótidos.
Las tecnologías modernas de secuenciación pueden generar un gran número de lecturas de secuencias en un solo experimento. Sin embargo, ninguna tecnología de secuenciación es perfecta, y cada instrumento generará diferentes tipos y cantidad de errores, como la llamada incorrecta de nucleótidos. Estas bases mal llamadas se deben a las limitaciones técnicas de cada plataforma de secuenciación.
Por lo tanto, es necesario comprender, identificar y excluir los tipos de error que pueden afectar a la interpretación de los análisis posteriores. El control de calidad de la secuencia es, por tanto, un primer paso esencial en su análisis. Detectar los errores a tiempo ahorra tiempo más adelante.
Crea un nuevo historial para este tutorial y dale un nombre apropiado
Haz click sobre el icono new-history en la parte superior del panel de historiales.
Haz clic sobre Unnamed history (o el nombre que tenga el historial sobre el que estás trabajando) (Haz clic para cambiar el nombre del historial) en la parte superior de tu panel de historial
Escribe el nombre nuevo
Pulsa Enter
Importe el archivo female_oral2.fastq-4143.gz de Zenodo o de la biblioteca de datos (pregunte a su instructor) Esta es una muestra de microbioma de una serpiente Jacques et al. 2021.
Abre el manejador de carga de datos de Galaxy (galaxy-upload (Upload) en la parte superior derecha del panel de herramientas)
Selecciona ‘Pegar/Traer datos’ Paste/Fetch Data
Copia los enlaces en el campo de textos
Presiona ‘Iniciar’ Start
Close Cierra la ventana.
Galaxy utiliza los URLs como nombres de forma predeterminada , así que los tendrás que cambiar a algunos que sean más útiles o informativos.
Como alternativa a cargar los datos desde una URL o desde su ordenador, los archivos también pueden estar disponibles desde una biblioteca de datos compartidos:
Entra en Libraries (panel izquierdo)
Navega a la carpeta correcta indicada por su instructor. - En la mayoría de los tutoriales de Galaxias los datos serán proporcionados en una carpeta llamada GTN - Material –> Topic name -> Tutorial name.
Seleccione los archivos deseados
Haz clic en Add to Historygalaxy-dropdown cerca de la parte superior y selecciona as Datasets en el menú desplegable
En la ventana emergente, elige
“Seleccionar historial “: el historial al que desea importar los datos (o crear uno nuevo)
Haga clic en Import
Cambie el nombre del conjunto de datos importado a Reads.
Acabamos de importar un archivo a Galaxy. Este archivo es similar a los datos que podríamos obtener directamente de una instalación de secuenciación: un archivo FASTQ.
Práctica: Inspeccione el archivo FASTQ
Inspeccione el archivo haciendo clic en el galaxy-eye (ojo)
Aunque parece complicado (y puede que lo sea), el formato FASTQ es fácil de entender con un poco de decodificación.
Cada lectura, que representa un fragmento de la biblioteca, está codificada por 4 líneas:
Línea
Descripción
1
Empieza siempre con un @ seguido de la información de la lecture
2
La secuenciación de nucleótidos
3
Empieza siempre con un + y contiene a veces la misma información qu ela línea 1
4
Contiene una cadena de caracteres que representa las puntuaciones de calidad asociadas a cada base de la secuencia nucleotídica; debe tener el mismo número de caracteres que la línea 2.
Así, por ejemplo, la primera secuencia en nuestro archivo es:
Significa que el fragmento llamado @M00970 corresponde a la secuencia de ADN GTGCCAGCCGCCGCGGTAGTCCGACGTGGCTGTCTCTTATACACATCTCCGAGCCCACGAGACCGAAGAACATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAAAAAAACAAAAAAAAAAAAAGAAGCAAATGACGATTCAAGAAAGAAAAAAACACAGAATACTAACAATAAGTCATAAACATCATCAACATAAAAAAGGAAATACACTTACAACACATATCAATATCTAAAATAAATGATCAGCACACAACATGACGATTACCACACATGTGTACTACAAGTCAACTA y esta secuencia ha sido secuenciada con una calidad GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGFGGGGGGAFFGGFGGGGGGGGFGGGGGGGGGGGGGGFGGG+38+35*311*6,,31=******441+++0+0++0+*1*2++2++0*+*2*02*/***1*+++0+0++38++00++++++++++0+0+2++*+*+*+*+*****+0**+0**+***+)*.***1**//*)***)/)*)))*)))*),)0(((-((((-.(4(,,))).,(())))))).)))))))-))-(.
¿Pero qué significa esta puntuación de calidad?
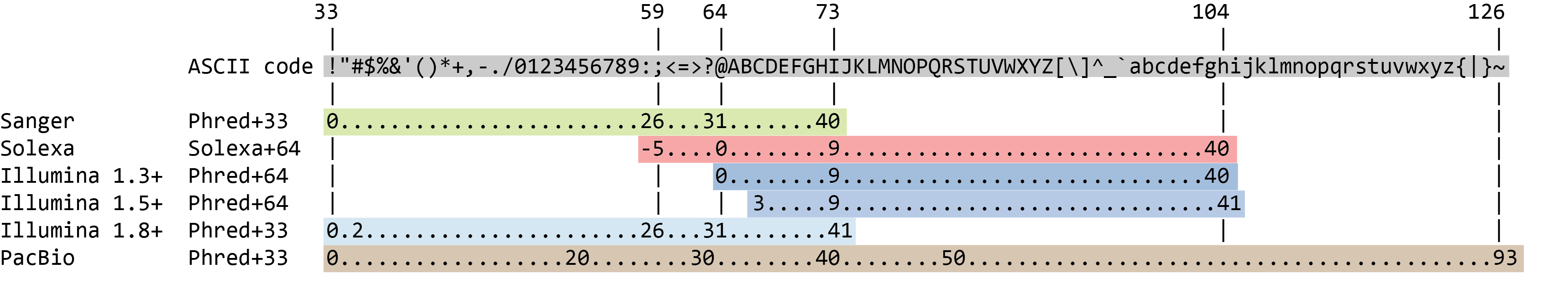
La puntuación de calidad de cada secuencia es una cadena de caracteres, uno por cada base de la secuencia de nucleótidos, que se utiliza para caracterizar la probabilidad de identificación errónea de cada base. La puntuación se codifica utilizando la tabla de caracteres ASCII (con algunas diferencias históricas):
Para ahorrar espacio, el secuenciador registra un carácter ASCII para representar las puntuaciones 0-42. Por ejemplo, 10 corresponde a “+” y 40 a “I”. FastQC sabe cómo traducir esto. A menudo se denomina puntuación “Phred”.
Así que hay un carácter ASCII asociado a cada nucleótido, que representa su puntuación de calidad Phred, la probabilidad de una llamada de base incorrecta:
Puntuación de calidad Phred
Probabilidad de una llamada de base incorrecta
Precisión de llamada de base
10
1 in 10
90%
20
1 in 100
99%
30
1 in 1000
99.9%
40
1 in 10,000
99.99%
50
1 in 100,000
99.999%
60
1 in 1,000,000
99.9999%
¿Qué representa 0-42? Estos números, cuando se introducen en una fórmula, nos indican la probabilidad de error para esa base. Esta es la fórmula, donde Q es nuestra puntuación de calidad (0-42) y P es la probabilidad de error:
Q = -10 log10(P)
Utilizando esta fórmula, podemos calcular que una puntuación de calidad de 40 significa sólo 0,00010 de probabilidad de error
Preguntas
¿Qué carácter ASCII corresponde a la peor puntuación Phred para Illumina 1.8+?
¿Cuál es la puntuación de calidad Phred del 3er nucleótido de la 1ª secuencia?
¿Cómo calcular la precisión de la base de nucleótidos con el código ASCII /?
¿Cuál es la precisión de este 3er nucleótido?
La peor puntuación Phred es la más pequeña, por lo tanto 0. Para Illumina 1.8+, corresponde al carácter !.
El 3er nucleótido de la 1ª secuencia tiene un carácter ASCII G, que corresponde a una puntuación de 38.
Se puede calcular de la siguiente manera:
El código ASCII para / es 47
Puntuación de calidad = 47-33=14
Fórmula para hallar la probabilidad de error: \(P = 10^-Q/10})
Probabilidad de error = \(10^{-14/10}\) = 0.03981 = 3.981 %
Por lo tanto Exactitud = 100 - 3.981 = 96.019%
El nucleótido correspondiente G tiene una precisión de casi el 96%
Comentario
El lllumina actual (1.8+) utiliza el formato Sanger (Phred+33). Si está trabajando con conjuntos de datos más antiguos puede encontrarse con los esquemas de puntuación más antiguos. FastQCtool, una herramienta que utilizaremos más adelante en este tutorial, se puede utilizar para tratar de determinar qué tipo de codificación de calidad se utiliza (a través de la evaluación de la gama de valores Phred visto en el FASTQ).
Al mirar el archivo en Galaxy, parece que la mayoría de los nucleótidos tienen una puntuación alta (G correspondiente a una puntuación 38). ¿Es esto cierto para todas las secuencias? ¿Y a lo largo de toda la secuencia?
Evaluar la calidad con FASTQE 🧬😎 - sólo lecturas cortas
Para echar un vistazo a la calidad de la secuencia a lo largo de todas las secuencias, podemos utilizar FASTQE. Es una herramienta de código abierto que proporciona una forma sencilla y divertida de controlar la calidad de los datos de secuencias en bruto e imprimirlos como emoji. Puede utilizarla para hacerse una idea rápida de si sus datos presentan algún problema del que deba ser consciente antes de realizar cualquier otro análisis.
Práctica: Comprobación de calidad
FASTQE ( Galaxy version 0.3.1+galaxy0) con los siguientes parámetros
param-files“FastQ data “: Reads
param-select“Tipos de puntuación a mostrar “: Mean
Inspeccione el archivo HTML generado
En lugar de analizar las puntuaciones de calidad de cada lectura individual, FASTQE analiza la calidad de forma colectiva en todas las lecturas de una muestra y puede calcular la media de cada posición de nucleótido a lo largo de la longitud de las lecturas. A continuación se muestran los valores medios de este conjunto de datos.
Puede ver la puntuación de cada emoji en la documentación de fastqe. Los emojis de abajo, con puntuaciones Phred inferiores a 20, son los que esperamos no ver mucho.
Phred Quality Score
ASCII code
Emoji
0
!
🚫
1
”
❌
2
#
👺
3
$
💔
4
%
🙅
5
&
👾
6
’
👿
7
(
💀
8
)
👻
9
*
🙈
10
+
🙉
11
,
🙊
12
-
🐵
13
.
😿
14
/
😾
15
0
🙀
16
1
💣
17
2
🔥
18
3
😡
19
4
💩
Preguntas
¿Cuál es la puntuación media más baja en este conjunto de datos?
La puntuación más baja en este conjunto de datos es 😿 13.
Evaluar la calidad con FastQC - lecturas cortas y largas
Una forma adicional o alternativa de comprobar la calidad de la secuencia es con FastQC. Proporciona un conjunto modular de análisis que podemos utilizar para comprobar si nuestros datos tienen algún problema del que debamos ser conscientes antes de realizar cualquier otro análisis. Podemos utilizarlo, por ejemplo, para evaluar si hay adaptadores conocidos presentes en los datos. Lo ejecutaremos en el archivo FASTQ
Práctica: Comprobación de calidad
FASTQC ( Galaxy version 0.73+galaxy0) con los siguientes parámetros
param-files“Datos de lectura crudos de su historia actual “: Reads
Inspeccione el archivo HTML generado
Preguntas
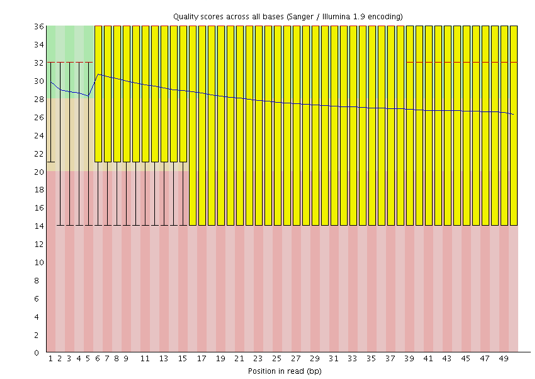
¿Qué codificación Phred se utiliza en el archivo FASTQ para estas secuencias?
Las puntuaciones Phred se codifican utilizando Sanger / Illumina 1.9 (Encoding en la tabla superior).
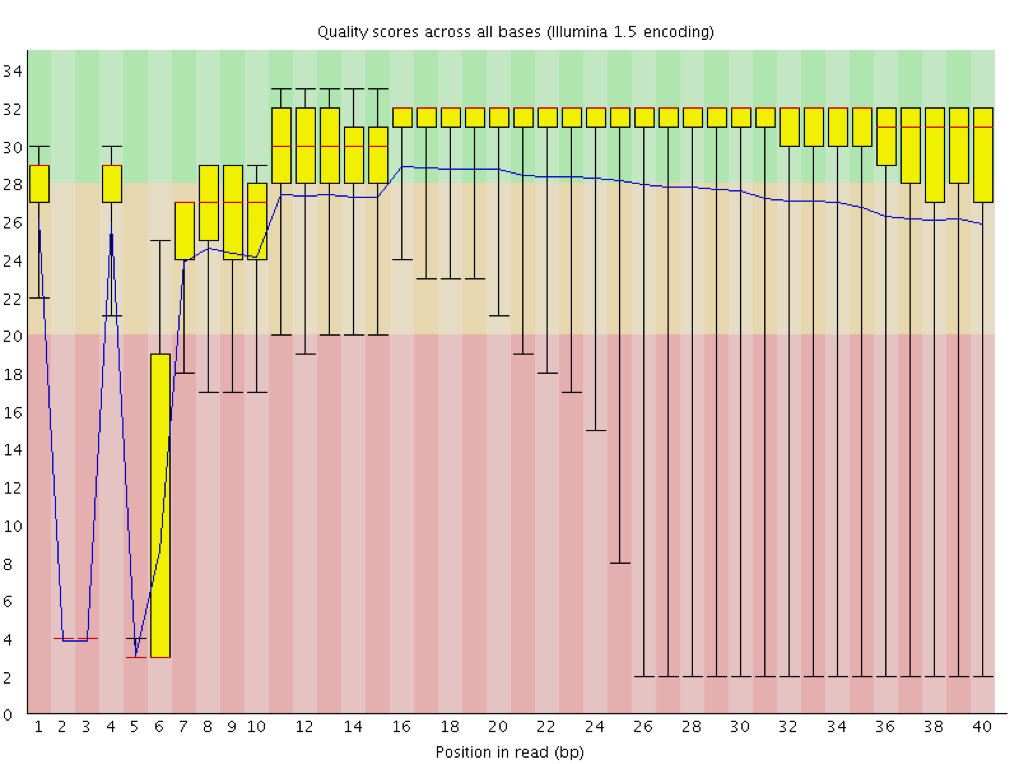
Calidad de la secuencia por base
Con FastQC podemos utilizar el gráfico de calidad de secuencia por base para comprobar la calidad de base de las lecturas, de forma similar a lo que hicimos con FASTQE.
En el eje x se muestra la posición de la base en la lectura. En este ejemplo, la muestra contiene lecturas de hasta 296bp de longitud.
El eje x no siempre es uniforme. Cuando se tienen lecturas largas, se aplica cierto binning para mantener las cosas compactas. Podemos verlo en nuestra muestra. Comienza con 1-10 bases individuales. Después, las bases se agrupan en una ventana de un cierto número de bases de ancho. Agrupar datos significa agrupar y es una técnica de preprocesamiento de datos utilizada para reducir los efectos de pequeños errores de observación. El número de posiciones de bases agrupadas depende de la longitud de la lectura. Con lecturas de más de 50bp, la última parte del gráfico mostrará estadísticas agregadas para ventanas de 5bp. Las lecturas más cortas tendrán ventanas más pequeñas y las más largas ventanas más grandes. El agrupamiento puede eliminarse al ejecutar FastQC estableciendo el parámetro “Disable grouping of bases for reads >50bp” en Yes.
Para cada posición, se dibuja un boxplot con:
el valor mediano, representado por la línea roja central
el rango intercuartílico (25-75%), representado por el recuadro amarillo
los valores 10% y 90% en los bigotes superior e inferior
la calidad media, representada por la línea azul
El eje y muestra las puntuaciones de calidad. Cuanto más alta es la puntuación, mejor es la llamada de la base. El fondo del gráfico divide el eje y en puntuaciones de muy buena calidad (verde), puntuaciones de calidad razonable (naranja) y lecturas de mala calidad (rojo).
Es normal en todos los secuenciadores Illumina que la mediana de la puntuación de calidad comience siendo más baja en las primeras 5-7 bases y luego aumente. La calidad de las lecturas en la mayoría de las plataformas disminuirá al final de la lectura. Esto se debe a menudo a la decadencia de la señal o al desfase durante el proceso de secuenciación. Los recientes avances en la química aplicada a la secuenciación han mejorado algo este aspecto, pero las lecturas son ahora más largas que nunca.
Decaimiento de la señal
La intensidad de la señal fluorescente decae con cada ciclo del proceso de secuenciación. Debido a la degradación de los fluoróforos, una proporción de las hebras del cluster no se está elongando. La proporción de la señal que se emite sigue disminuyendo con cada ciclo, lo que produce una disminución de las puntuaciones de calidad en el extremo 3’ de la lectura.
Fases
La señal empieza a difuminarse con el aumento del número de ciclos porque el cluster pierde sincronicidad. A medida que avanzan los ciclos, en algunas hebras se producen fallos aleatorios de nucleótidos a incorporar debido a:
Eliminación incompleta de los terminadores 3’ y fluoróforos
Incorporación de nucleótidos sin terminadores 3’ efectivos
Esto conduce a una disminución de las puntuaciones de calidad en el extremo 3’ de la lectura.
Estos son algunos perfiles de calidad de secuencias por base que pueden indicar problemas con la secuenciación.
Sobreagrupamiento
Las instalaciones de secuenciación pueden agrupar en exceso las celdas de flujo. El resultado son pequeñas distancias entre los clusters y un solapamiento de las señales. Dos clusters pueden interpretarse como un único cluster, detectándose señales fluorescentes mezcladas, lo que disminuye la pureza de la señal. Genera puntuaciones de calidad más bajas en toda la lectura.
Desglose de la instrumentación
Ocasionalmente pueden producirse algunos problemas con los instrumentos de secuenciación durante un experimento. Cualquier caída repentina en la calidad o un gran porcentaje de lecturas de baja calidad a lo largo de la lectura podría indicar un problema en la instalación. Algunos ejemplos de estos problemas:
Ráfaga múltiple
Pérdida de ciclos
Fallo de lectura 2
Con estos datos, debe ponerse en contacto con el centro de secuenciación. A menudo, es necesaria una resecuenciación (que, según nuestra experiencia, también ofrece la empresa).
Preguntas
¿Cómo cambia la puntuación media de calidad a lo largo de la secuencia?
¿Se observa esta tendencia en todas las secuencias?
La puntuación media de calidad (línea azul) desciende aproximadamente a mitad de estas secuencias. Es habitual que la calidad media descienda hacia el final de las secuencias, ya que los secuenciadores incorporan más nucleótidos incorrectos al final. Sin embargo, en esta muestra se observa un descenso muy acusado de la calidad a partir de la mitad.
Los gráficos de caja son cada vez más amplios a partir de la posición ~100. Significa que la puntuación de muchas secuencias cae desde la mitad de la secuencia. Después de 100 nucleótidos, más del 10% de las secuencias tienen puntuaciones por debajo de 20.
Cuando la mediana de calidad está por debajo de una puntuación Phred de ~20, deberíamos considerar la posibilidad de recortar las bases de mala calidad de la secuencia. Explicaremos este proceso en la sección Recortar y filtrar.
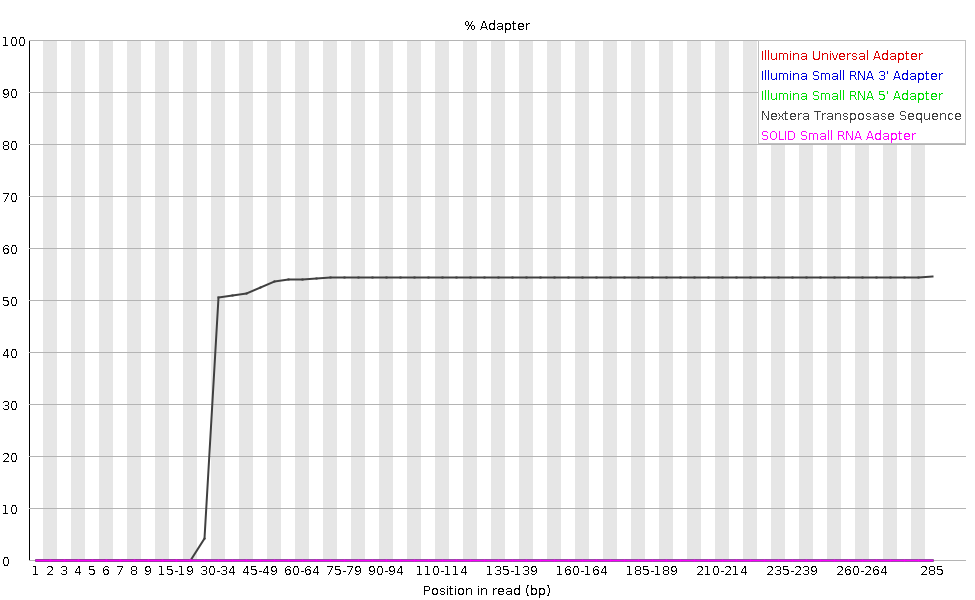
El gráfico muestra el porcentaje acumulado de lecturas con las diferentes secuencias adaptadoras en cada posición. Una vez que una secuencia adaptadora se ve en una lectura, se cuenta como presente hasta el final de la lectura, por lo que el porcentaje aumenta con la longitud de la lectura. FastQC puede detectar algunos adaptadores por defecto (p.ej. Illumina, Nextera), para otros podemos proporcionar un archivo de contaminantes como entrada a la herramienta FastQC.
Lo ideal sería que los datos de secuencia Illumina no tuvieran ninguna secuencia adaptadora. Pero con lecturas largas, algunas de las inserciones de la biblioteca son más cortas que la longitud de la lectura, lo que da lugar a una lectura a través del adaptador en el extremo 3’ de la lectura. Esta muestra de microbioma tiene lecturas relativamente largas y podemos ver que se ha detectado el adaptador Nextera.
El contenido de adaptadores también puede detectarse con bibliotecas RNA-Seq en las que la distribución de los tamaños de las inserciones de la biblioteca es variada y es probable que incluya algunas inserciones cortas.
Podemos ejecutar una herramienta de recorte como Cutadapt para eliminar este adaptador. Explicaremos este proceso en la sección de filtrado y recorte.
Las siguientes secciones detallan algunos de los otros gráficos generados por FastQC. Tenga en cuenta que algunos gráficos/módulos pueden dar advertencias, pero son normales para el tipo de datos con los que está trabajando, como se explica a continuación y en el FAQ de FASTQC. Los otros gráficos nos dan información para entender más profundamente la calidad de los datos, y para ver si se pueden hacer cambios en el laboratorio para obtener datos de mayor calidad en el futuro. Estas secciones son opcionales, y si desea saltárselas puede hacerlo:
Vaya directamente a la siguiente sección para aprender sobre el recorte de datos de extremos pareados
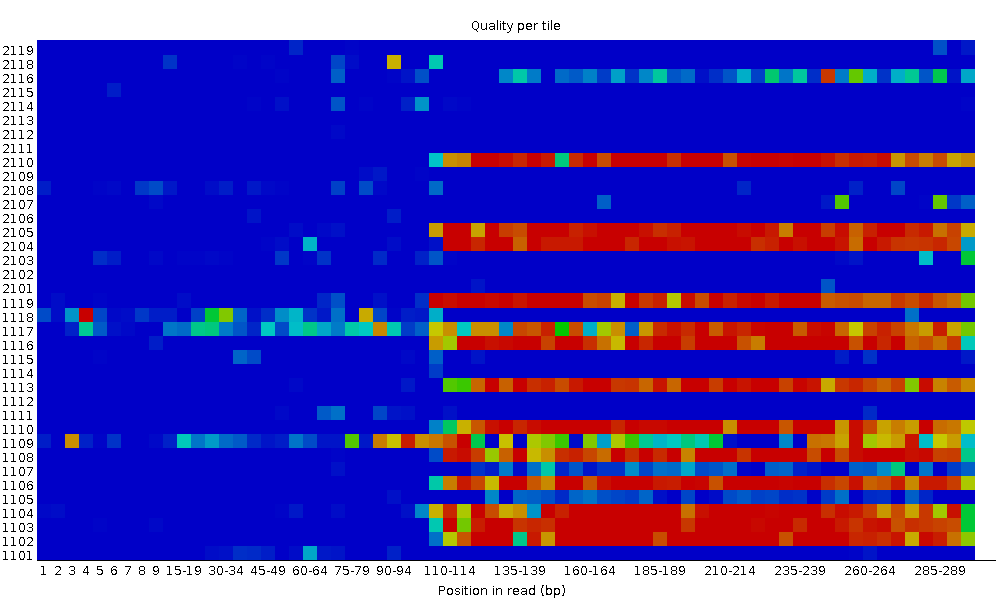
Calidad de la secuencia por mosaico
Este gráfico le permite observar las puntuaciones de calidad de cada mosaico en todas sus bases para ver si hubo una pérdida de calidad asociada a una sola parte de la celda de flujo. El gráfico muestra la desviación de la calidad media de cada mosaico de la célula de flujo. Los colores más vivos indican que las lecturas en el mosaico dado tienen peores calidades para esa posición que las lecturas en otros mosaicos. Con esta muestra, se puede ver que ciertos mosaicos muestran una calidad consistentemente pobre, especialmente a partir de ~100bp. Un buen gráfico debería ser azul en todas partes.
Este gráfico sólo aparecerá para las bibliotecas Illumina que conserven sus identificadores de secuencia originales. En ellos está codificada la celda de flujo de la que procede cada lectura.
En algunos casos, los productos químicos utilizados durante la secuenciación se agotan un poco con el tiempo y los últimos mosaicos tienen peores productos químicos, lo que hace que las reacciones de secuenciación sean un poco propensas a errores. El gráfico “Calidad de la secuencia por mosaico” tendrá entonces algunas líneas horizontales como esta:
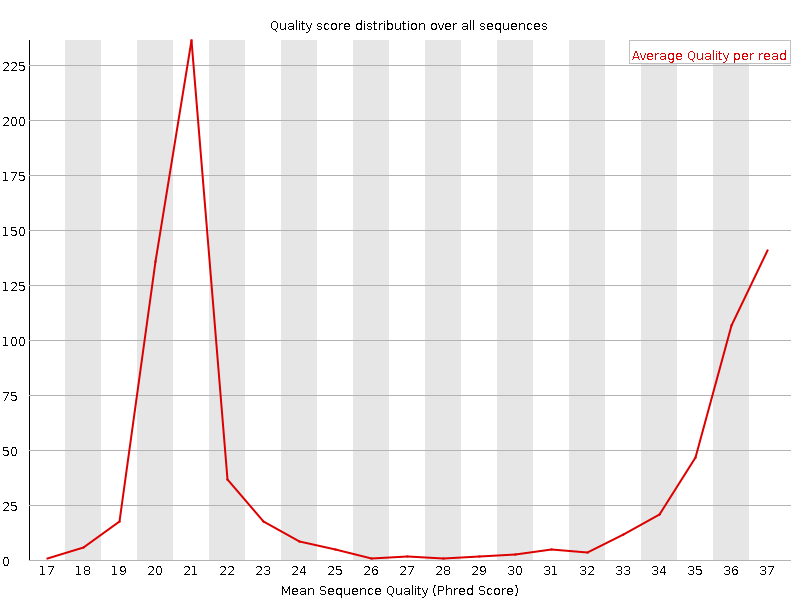
Puntuaciones de calidad por secuencia
Traza la puntuación de calidad media sobre la longitud total de todas las lecturas en el eje x y da el número total de lecturas con esta puntuación en el eje y:
La distribución de la calidad media de lectura debe ser un pico agudo en el rango superior de la parcela. También puede informar si un subconjunto de las secuencias tiene valores de calidad universalmente bajos: puede ocurrir porque algunas secuencias tienen imágenes pobres (en el borde del campo de visión, etc.), sin embargo, éstas deberían representar sólo un pequeño porcentaje del total de secuencias.
Figura 6: Contenido por secuencia de bases para una biblioteca de ADN
“Per Base Sequence Content” muestra el porcentaje de cada uno de los cuatro nucleótidos (T, C, A, G) en cada posición de todas las lecturas del archivo de secuencia de entrada. Como en el caso de la calidad de la secuencia por base, el eje x no es uniforme.
En una biblioteca aleatoria esperaríamos que hubiera poca o ninguna diferencia entre las cuatro bases. La proporción de cada una de las cuatro bases debería permanecer relativamente constante a lo largo de la lectura con %A=%T y %G=%C, y las líneas de este gráfico deberían ser paralelas entre sí. Estos son datos de amplicón, donde el ADN 16S es amplificado por PCR y secuenciado, por lo que esperaríamos que este gráfico tuviera algún sesgo y no mostrara una distribución aleatoria.
Cabe señalar que algunos tipos de bibliotecas siempre producirán una composición sesgada de la secuencia, normalmente al inicio de la lectura. Las bibliotecas producidas por cebado usando hexámeros aleatorios (incluyendo casi todas las bibliotecas RNA-Seq), y aquellas que fueron fragmentadas usando transposasas, contendrán un sesgo intrínseco en las posiciones en las que comienzan las lecturas (las primeras 10-12 bases). Este sesgo no implica una secuencia específica, sino que proporciona un enriquecimiento de una serie de diferentes K-mers en el extremo 5’ de las lecturas. Aunque se trata de un verdadero sesgo técnico, no es algo que pueda corregirse recortando y, en la mayoría de los casos, no parece afectar negativamente al análisis posterior. Sin embargo, producirá una advertencia o un error en este módulo.
Los datos ChIP-seq también pueden encontrar sesgos en la secuencia de inicio de lectura en este gráfico si se fragmentan con transposasas. Con datos convertidos por bisulfito, por ejemplo datos HiC, se espera una separación de G de C y A de T:
Al final, hay un cambio general en la composición de la secuencia. Si el desplazamiento se correlaciona con una pérdida de calidad de la secuenciación, se puede sospechar que los errores se producen con un sesgo de secuencia más uniforme que las bibliotecas convertidas por bisulfito. El recorte de las secuencias solucionó este problema, pero si no se hubiera hecho habría tenido un efecto dramático en las llamadas de metilación que se hicieron.
Preguntas
¿Por qué hay una advertencia para los gráficos de contenido de secuencia por base?
Al principio de las secuencias, el contenido de secuencia por base no es realmente bueno y los porcentajes no son iguales, como se espera para datos de amplicón 16S.
Este gráfico muestra el número de lecturas frente al porcentaje de bases G y C por lectura. Se compara con una distribución teórica que asume un contenido uniforme de GC para todas las lecturas, esperado para la secuenciación de escopeta del genoma completo, donde el pico central corresponde al contenido general de GC del genoma subyacente. Dado que no se conoce el contenido de GC del genoma, se calcula el contenido modal de GC a partir de los datos observados y se utiliza para construir una distribución de referencia.
Una distribución con forma inusual podría indicar una biblioteca contaminada o algún otro tipo de subconjunto sesgado. Una distribución normal desplazada indica algún sesgo sistemático, que es independiente de la posición de las bases. Si hay un sesgo sistemático que crea una distribución normal desplazada, el módulo no lo marcará como error, ya que no sabe cuál debería ser el contenido de GC de su genoma.
Pero también hay otras situaciones en las que puede producirse una distribución de forma inusual. Por ejemplo, con la secuenciación de ARN puede haber una distribución mayor o menor del contenido medio de GC entre los transcritos, lo que hace que el gráfico observado sea más ancho o más estrecho que una distribución normal ideal.
Preguntas
¿Por qué fallan los gráficos de contenido de GC por secuencia?
Hay múltiples picos. Esto puede ser indicativo de contaminación inesperada, como adaptador, ARNr o secuencias sobrerrepresentadas. O puede ser normal si se trata de datos de amplicón o si tiene transcritos de ARN-seq muy abundantes.
Distribución de la longitud de la secuencia
Este gráfico muestra la distribución de tamaños de fragmentos en el archivo analizado. En muchos casos, esto producirá un simple gráfico que mostrará un pico en un solo tamaño, pero para los archivos FASTQ de longitud variable mostrará las cantidades relativas de cada tamaño diferente de fragmento de secuencia. Nuestro gráfico muestra la longitud variable a medida que recortamos los datos. El pico más grande está a 296bp, pero hay un segundo pico grande a ~100bp. Así que, aunque nuestras secuencias tienen una longitud de hasta 296bp, muchas de las secuencias de buena calidad son más cortas. Esto se corresponde con la caída que observamos en la calidad de la secuencia a ~100bp y las rayas rojas que comienzan en esta posición en el gráfico de calidad de la secuencia por mosaico.
Figura 8: Distribución de la longitud de secuencia
Algunos secuenciadores de alto rendimiento generan fragmentos de secuencias de longitud uniforme, pero otros pueden contener lecturas de longitudes muy variables. Incluso dentro de las bibliotecas de longitud uniforme, algunas pipelines recortarán las secuencias para eliminar las llamadas de bases de baja calidad del final o de las primeras \(n\) bases si coinciden con las primeras \(n\) bases del adaptador hasta un 90% (por defecto), con a veces \(n = 1\).
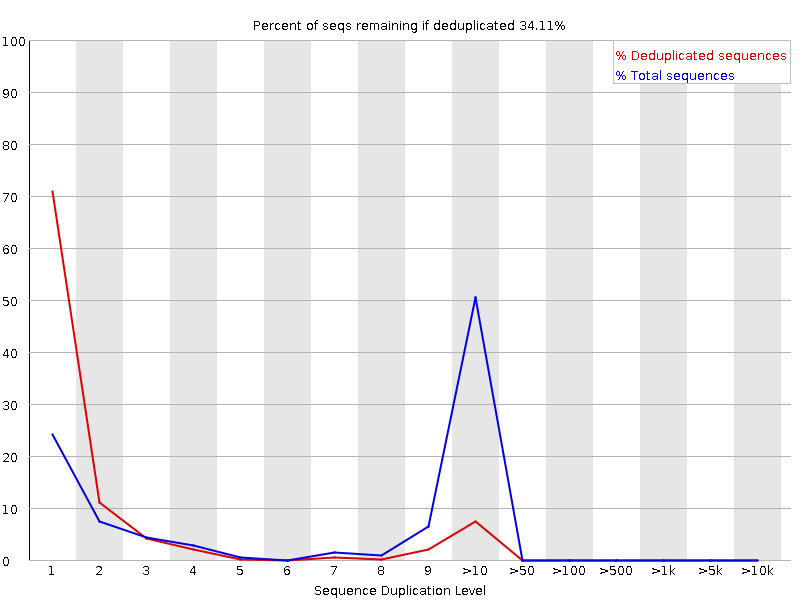
Niveles de duplicación de secuencias
El gráfico muestra en azul el porcentaje de lecturas de una secuencia dada en el archivo que están presentes un número determinado de veces en el archivo:
En una biblioteca diversa, la mayoría de las secuencias aparecerán sólo una vez en el conjunto final. Un nivel bajo de duplicación puede indicar un nivel muy alto de cobertura de la secuencia objetivo, pero un nivel alto de duplicación es más probable que indique algún tipo de sesgo de enriquecimiento.
Se pueden encontrar dos fuentes de lecturas duplicadas:
Duplicación PCR en la que los fragmentos de la biblioteca se han sobre-representado debido a un enriquecimiento PCR sesgado
Es preocupante porque los duplicados de PCR falsean la proporción real de secuencias en la entrada.
Secuencias verdaderamente sobrerrepresentadas, como transcritos muy abundantes en una biblioteca de RNA-Seq o en datos de amplicón (como esta muestra)
Es un caso esperado y no es preocupante porque representa fielmente la entrada.
FastQC cuenta el grado de duplicación de cada secuencia en una biblioteca y crea un gráfico que muestra el número relativo de secuencias con diferentes grados de duplicación. Hay dos líneas en el gráfico:
Línea azul: distribución de los niveles de duplicación para el conjunto completo de secuencias
Línea roja: distribución para las secuencias deduplicadas con las proporciones del conjunto deduplicado que provienen de diferentes niveles de duplicación en los datos originales.
Para los datos shotgun de genoma completo se espera que casi el 100% de sus lecturas sean únicas (que aparezcan sólo una vez en los datos de la secuencia). La mayoría de las secuencias deberían situarse en el extremo izquierdo del gráfico, tanto en la línea roja como en la azul. Esto indica que la biblioteca es muy diversa y que no se ha secuenciado en exceso. Si la profundidad de secuenciación es extremadamente alta (por ejemplo, > 100 veces el tamaño del genoma) puede aparecer alguna duplicación inevitable de secuencias: en teoría, sólo hay un número finito de lecturas de secuencias completamente únicas que pueden obtenerse a partir de cualquier muestra de ADN de entrada.
Los enriquecimientos más específicos de subconjuntos o la presencia de contaminantes de baja complejidad tenderán a producir picos hacia la derecha del gráfico. Estos picos de alta duplicación aparecerán con mayor frecuencia en el trazo azul, ya que constituyen una proporción elevada de la biblioteca original, pero suelen desaparecer en el trazo rojo, ya que constituyen una proporción insignificante del conjunto deduplicado. Si los picos persisten en el trazo rojo, esto sugiere que hay un gran número de secuencias diferentes altamente duplicadas, lo que podría indicar un conjunto contaminante o una duplicación técnica muy grave.
Suele ocurrir en la secuenciación de ARN que hay transcritos muy abundantes y otros poco abundantes. Es de esperar que se observen lecturas duplicadas para los transcritos de alta abundancia:
Secuencias sobrerrepresentadas
Una biblioteca normal de alto rendimiento contendrá un conjunto diverso de secuencias, sin que ninguna secuencia individual represente una pequeña fracción del conjunto. Descubrir que una única secuencia está muy sobrerrepresentada en el conjunto significa que es altamente significativa desde el punto de vista biológico o indica que la biblioteca está contaminada o no es tan diversa como se esperaba.
FastQC enumera todas las secuencias que representan más del 0,1% del total. Para cada secuencia sobrerrepresentada, FastQC buscará coincidencias en una base de datos de contaminantes comunes e informará de la mejor coincidencia que encuentre. Las coincidencias deben tener al menos 20bp de longitud y no más de 1 desajuste. Encontrar una coincidencia no significa necesariamente que ésta sea la fuente de la contaminación, pero puede orientarle en la dirección correcta. También hay que tener en cuenta que muchas secuencias adaptadoras son muy similares entre sí, por lo que es posible que obtenga una coincidencia que no sea técnicamente correcta, pero que tenga una secuencia muy similar a la coincidencia real.
Los datos de secuenciación de ARN pueden tener algunas transcripciones que son tan abundantes que se registran como secuencia sobrerrepresentada. Con los datos de secuenciación de ADN, ninguna secuencia debería estar presente con una frecuencia lo suficientemente alta como para aparecer en la lista, pero a veces podemos ver un pequeño porcentaje de lecturas adaptadoras.
Preguntas
¿Cómo podríamos averiguar cuáles son las secuencias sobrerrepresentadas?
Podemos hacer BLAST con las secuencias sobrerrepresentadas para ver cuáles son. En este caso, si tomamos la secuencia más sobrerrepresentada
y usamos blastn contra la base de datos Nucleotide (nr/nt) por defecto no obtenemos ningún resultado. Pero si usamos VecScreen vemos que es el adaptador Nextera.
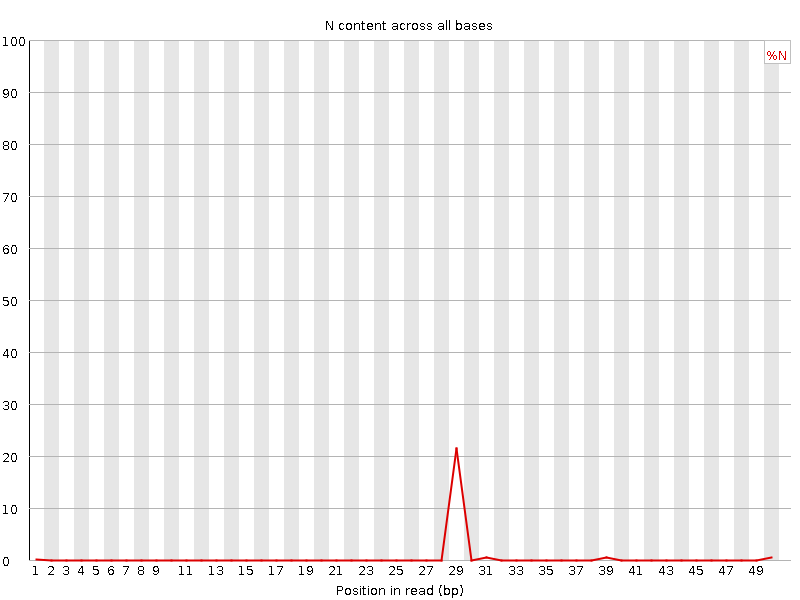
Si un secuenciador no puede realizar una llamada de base con suficiente confianza, escribirá una “N” en lugar de una llamada de base convencional. Este gráfico muestra el porcentaje de llamadas de base en cada posición o ubicación para las que se ha escrito una N.
No es inusual ver una proporción muy alta de Ns que aparecen en una secuencia, especialmente cerca del final de una secuencia. Pero esta curva nunca debería elevarse notablemente por encima de cero. Si lo hace, esto indica que se ha producido un problema durante la ejecución de la secuenciación. En el siguiente ejemplo, un error provocó que el instrumento no pudiera llamar una base en aproximadamente el 20% de las lecturas en la posición 29:
Contenido Kmer
Este gráfico no se muestra por defecto. Como se indica en el formulario de la herramienta, si desea utilizar este módulo, debe habilitarlo mediante un submódulo personalizado y un archivo de límites. Con este módulo, FastQC realiza un análisis genérico de todas las secuencias cortas de nucleótidos de longitud k (kmer, con k = 7 por defecto) comenzando en cada posición a lo largo de la lectura en la biblioteca para encontrar aquellas que no tienen una cobertura uniforme a lo largo de la longitud de sus lecturas. Cualquier kmer dado debe estar representado uniformemente en toda la longitud de la lectura.
FastQC informará de la lista de kmers que aparecen en posiciones específicas con una frecuencia mayor de la esperada. Esto puede deberse a diferentes fuentes de sesgo en la biblioteca, incluida la presencia de secuencias adaptadoras de lectura que se acumulan al final de las secuencias. La presencia de secuencias sobrerrepresentadas en la biblioteca (como dímeros de adaptador) hace que el gráfico de kmer esté dominado por el kmer de estas secuencias. Cualquier kmer sesgado debido a otros sesgos interesantes puede entonces diluirse y no ser fácil de ver.
El siguiente ejemplo es de una biblioteca DNA-Seq de alta calidad. Los kmers sesgados cerca del inicio de la lectura probablemente se deban a una ligera eficiencia dependiente de la secuencia del cizallamiento del ADN o a un resultado de cebado aleatorio:
Este módulo puede ser muy difícil de interpretar. El gráfico de contenido de adaptadores y la tabla de secuencias sobrerrepesentadas son más fáciles de interpretar y pueden darle suficiente información sin necesidad de este gráfico. Las bibliotecas RNA-seq pueden tener kmers altamente representados que derivan de secuencias altamente expresadas. Para obtener más información sobre este gráfico, consulte la FastQC Kmer Content documentation.
Intentamos explicar aquí diferentes informes de FastQC y algunos casos de uso. Puede encontrar más información sobre este tema y también sobre algunos problemas comunes de secuenciación de nueva generación en QCFAIL.com
ARN pequeño/micro
En las pequeñas bibliotecas de ARN, normalmente tenemos un conjunto relativamente pequeño de secuencias únicas y cortas. Las bibliotecas de ARN pequeño no se cizallan aleatoriamente antes de añadir adaptadores de secuenciación a sus extremos: todas las lecturas para clases específicas de microARN serán idénticas. El resultado será:
Contenido de secuencia por base extremadamente sesgado
Distribución extremadamente estrecha del contenido de GC
Niveles muy altos de duplicación de secuencias
Abundancia de secuencias sobrerrepresentadas
Lectura en adaptadores
Amplicón
Las bibliotecas de amplicones se preparan mediante la amplificación por PCR de una diana específica. Por ejemplo, la región hipervariable V4 del gen 16S rRNA bacteriano. Se espera que todas las lecturas de este tipo de bibliotecas sean casi idénticas. El resultado será:
Contenido de secuencia por base extremadamente sesgado
Distribución extremadamente estrecha del contenido de GC
Niveles muy altos de duplicación de secuencias
Abundancia de secuencias sobrerrepresentadas
Secuenciación por bisulfito o metilación
Con bisulfito o secuenciación por metilación, la mayoría de las bases de citosina (C) se convierten en timina (T). El resultado será:
Contenido sesgado de la secuencia por base
Contenido de GC sesgado por secuencia
Contaminación de dímeros adaptadores
Cualquier tipo de biblioteca puede contener un porcentaje muy pequeño de fragmentos dímeros de adaptador (es decir, sin inserción). Es más probable encontrarlos en bibliotecas de amplicones construidas totalmente por PCR (por formación de dímeros de cebadores PCR) que en bibliotecas DNA-Seq o RNA-Seq construidas por ligadura de adaptadores. Si una fracción suficiente de la biblioteca está formada por dímeros de adaptador, se notará en el informe FastQC:
Caída de la calidad de la secuencia por base después de la base 60
Posible distribución bimodal de las puntuaciones de calidad por secuencia
Patrón distinto observado en el contenido de la secuencia por base hasta la base 60
Pico en el contenido de GC por secuencia
Adaptador de coincidencia de secuencia sobrerrepresentada
Contenido de adaptador > 0% a partir de la base 1
Comentario: Secuencias de mala calidad
Si la calidad de las lecturas no es buena, siempre debemos comprobar primero qué es lo que está mal y pensar en ello: puede venir del tipo de secuenciación o de lo que secuenciamos (gran cantidad de secuencias sobrerrepresentadas en datos transcriptómicos, porcentaje sesgado de bases en datos HiC).
También puede preguntar en el centro de secuenciación, especialmente si la calidad es realmente mala: los tratamientos de calidad no pueden resolverlo todo. Si se cortan demasiadas bases de mala calidad, las lecturas correspondientes se filtrarán y se perderán.
Recorte y filtrado - lecturas cortas
La calidad desciende en el centro de estas secuencias. Esto podría causar sesgos en los análisis posteriores con estos nucleótidos potencialmente mal llamados. Las secuencias deben ser tratadas para reducir el sesgo en los análisis posteriores. El recorte puede ayudar a aumentar el número de lecturas que el alineador o el ensamblador son capaces de utilizar con éxito, reduciendo el número de lecturas que quedan sin mapear o ensamblar. En general, los tratamientos de calidad incluyen:
Recorte/corte/enmascaramiento de secuencias
de regiones de baja puntuación de calidad
principio/final de secuencia
eliminación de adaptadores
Filtrado de secuencias
con una puntuación media de calidad baja
demasiado corto
con demasiadas bases ambiguas (N)
Para llevar a cabo esta tarea utilizaremos CutadaptMarcel 2011, una herramienta que mejora la calidad de la secuencia automatizando el recorte de adaptadores así como el control de calidad. Lo haremos:
Recorte de bases de baja calidad de los extremos. El recorte de calidad se realiza antes de cualquier recorte de adaptador. Estableceremos el umbral de calidad en 20, un umbral de uso común, ver más en GATK’s Phred Score FAQ.
Recorte del adaptador con Cutadapt. Para ello necesitamos suministrar la secuencia del adaptador. En este ejemplo, Nextera es el adaptador detectado. Podemos encontrar la secuencia del adaptador Nextera en el sitio web de IlluminaCTGTCTCTTATACACATCT. Recortaremos esa secuencia del extremo 3’ de las lecturas.
Filtro de secuencias con longitud < 20 después del recorte
Práctica: Mejora de la calidad de la secuencia
Cutadapt ( Galaxy version 4.9+galaxy1) con los siguientes parámetros
"”Single-end or Paired-end reads?” “: Single-end
param-file“FASTQ/A file “: Reads (Conjunto de datos de entrada)
Si su archivo FASTQ no se puede seleccionar, puede comprobar si el formato es FASTQ con valores de calidad escalados por Sanger (fastqsanger.gz). Puede editar el tipo de datos haciendo clic en el símbolo del lápiz.
En “Read 1 Adapters “:
“1: 3’ (Fin) Adapters “:
“Source “: Enter custom sequence
“Custom 3’ adapter sequence”: CTGTCTCTTATACACATCT
En “Other Read Trimming Options “
“Quality cutoff(s) (R1) “: 20
En “Read Filtering Options “ (Opciones de filtrado de lectura)
“Minimum lenght (R1) “: 20
param-select“Additional outputs to generate “: Report
Inspeccione el archivo txt generado (Report)
Preguntas
¿Qué % de lecturas contienen adaptador?
¿Qué % de lecturas se han recortado por mala calidad?
¿Qué % de lecturas se han eliminado por ser demasiado cortas?
56.8% lecturas contienen adaptador (Reads with adapters:)
El 35,1% de las lecturas han sido recortadas por mala calidad (Quality-trimmed:)
0 % de lecturas eliminadas por ser demasiado cortas
Una de las mayores ventajas de Cutadapt en comparación con otras herramientas de recorte (¡por ejemplo TrimGalore!) es que tiene una buena documentación que explica cómo funciona la herramienta en detalle.
El algoritmo de recorte de calidad Cutadapt consta de tres sencillos pasos:
Reste el valor umbral elegido del valor de calidad de cada posición
Calcule una suma parcial de estas diferencias desde el final de la secuencia hasta cada posición (siempre que la suma parcial sea negativa)
Corte en el valor mínimo de la suma parcial
En el siguiente ejemplo, suponemos que el extremo 3’ debe ser de calidad recortada con un umbral de 10 y tenemos los siguientes valores de calidad
42 40 26 27 8 7 11 4 2 3
Restar el umbral
32 30 16 17 -2 -3 1 -6 -8 -7
Sume los números, empezando por el extremo 3’ (sumas parciales) y deténgase antes si la suma es mayor que cero
(70) (38) 8 -8 -25 -23 -20, -21 -15 -7
Los números entre paréntesis no se calculan (porque 8 es mayor que cero), pero se muestran aquí para completar.
Elija la posición del mínimo (-25) como posición de recorte
Por lo tanto, la lectura se recorta a las cuatro primeras bases, que tienen valores de calidad
42 40 26 27
Obsérvese que, por lo tanto, las posiciones con un valor de calidad superior al umbral elegido también se eliminan si están incrustadas en regiones de menor calidad (la suma parcial es decreciente si los valores de calidad son inferiores al umbral). La ventaja de este procedimiento es que es robusto frente a un pequeño número de posiciones con una calidad superior al umbral.
Las alternativas a este procedimiento serían:
Corte tras la primera posición con una calidad inferior al umbral
Enfoque de ventana deslizante
El enfoque de la ventana deslizante comprueba que la calidad media de cada ventana de secuencia de longitud especificada sea mayor que el umbral. Tenga en cuenta que, a diferencia del enfoque de cutadapt, este enfoque tiene un parámetro más y la robustez depende de la longitud de la ventana (en combinación con el umbral de calidad). Ambos enfoques están implementados en Trimmomatic.
Podemos examinar nuestros datos recortados con FASTQE y/o FastQC.
Práctica: Comprobación de la calidad tras el recorte
FASTQE ( Galaxy version 0.3.1+galaxy0): Vuelva a ejecutar FASTQE con los siguientes parámetros
param-files“FastQ data “: Cutadapt Read 1 Output
param-select“Score types to show “:: Mean
Inspeccione el nuevo informe FASTQE
Preguntas
Compare la salida FASTQE con la anterior antes del recorte anterior. ¿Ha mejorado la calidad de la secuencia?
Si deseas ver dos o más conjuntos de datos al mismo tiempo, puedes usar la función Scratchbook en Galaxy:
Haz clic en el icono Scratchbookgalaxy-scratchbook en la barra de menú superior.
Debería aparecer ver una pequeña marca de verificación en el icono
Vergalaxy-eye un conjunto de datos haciendo clic en el icono de ojo galaxy-eye para ver el resultado.
Deberías ver la salida en una ventana emergente sobre Galaxy
Puedes cambiar el tamaño de esta ventana arrastrando la esquina inferior derecha
Haz clic fuera del archivo para salir del Scratchbook
Vergalaxy-eye un segundo conjunto de datos de tu historial
Ahora deberías poder ver una segunda ventana con el nuevo conjunto de datos
Esto hace que sea más fácil comparar las dos salidas.
Repite estos pasos para todos los archivos que desees comparar.
Puedes desactivar Scratchbookgalaxy-scratchbook haciendo clic en el icono nuevamente.
Sí, los emojis de puntuación de calidad se ven mejor (más felices) ahora.
Con FastQC podemos ver que mejoramos la calidad de las bases en el conjunto de datos y eliminamos el adaptador.
Tenemos algunas rayas rojas porque hemos recortado esas regiones de las lecturas.
Ahora tenemos un pico de alta calidad en lugar de uno de alta y otro de baja calidad que teníamos anteriormente.
No tenemos la misma representación de las bases que antes, ya que se trata de datos de amplicones.
Ahora tenemos un único pico principal de GC debido a la eliminación del adaptador.
Esto es lo mismo que antes, ya que no tenemos Ns en estas lecturas.
Ahora tenemos múltiples picos y un rango de longitudes, en lugar del único pico que teníamos antes del recorte cuando todas las secuencias tenían la misma longitud.
Preguntas
¿A qué corresponde la secuencia más sobrerrepresentada GTGTCAGCCGCCGCGGTAGTCCGACGTGG?
Si tomamos la secuencia más sobrerrepresentada
>overrep_seq1_after
GTGTCAGCCGCCGCGGTAGTCCGACGTGG
y usamos blastn contra la base de datos por defecto Nucleotide (nr/nt) vemos que los resultados más altos son para genes 16S rRNA. Esto tiene sentido, ya que se trata de datos de amplicones 16S, en los que el gen 16S se amplifica por PCR.
Procesamiento de múltiples conjuntos de datos
Procesamiento de datos por pares
En la secuenciación por pares, los fragmentos se secuencian por ambos lados. Este enfoque da lugar a dos lecturas por fragmento, con la primera lectura orientada hacia delante y la segunda hacia atrás. Con esta técnica, tenemos la ventaja de obtener más información sobre cada fragmento de ADN en comparación con las lecturas secuenciadas únicamente por secuenciación de un solo extremo:
Se conoce la distancia entre ambas lecturas y, por lo tanto, es información adicional que puede mejorar el mapeo de lecturas.
La secuenciación por pares genera 2 archivos FASTQ:
Un archivo con las secuencias correspondientes a la orientación hacia adelante de todos los fragmentos
Un archivo con las secuencias correspondientes a la orientación inversa de todos los fragmentos
Normalmente reconocemos estos dos archivos que pertenecen a una muestra por el nombre que tiene el mismo identificador para las lecturas pero una extensión diferente, por ejemplo sampleA_R1.fastq para las lecturas hacia adelante y sampleA_R2.fastq para las lecturas hacia atrás. También puede ser _f o _1 para las lecturas directas y _r o _2 para las lecturas inversas.
Los datos que analizamos en el paso anterior eran datos de extremo único, por lo que importaremos un conjunto de datos de ARN-seq de extremo pareado. Ejecutaremos FastQC y agregaremos los dos informes con MultiQC Ewels et al. 2016.
Práctica: Evaluación de la calidad de las lecturas paired-end
Importe las lecturas pareadas GSM461178_untreat_paired_subset_1.fastq y GSM461178_untreat_paired_subset_2.fastq de Zenodo o de la biblioteca de datos (pregunte a su instructor)
FASTQC ( Galaxy version 0.73+galaxy0) con ambos conjuntos de datos:
param-files“Raw read data from your current history “: ambos conjuntos de datos cargados.
Haga clic en param-filesMultiple datasets
Selecciona varios archivos manteniendo pulsada la tecla Ctrl (o COMMAND) y haciendo clic en los archivos que te interesen
MultiQC ( Galaxy version 1.9+galaxy1) con los siguientes parámetros para agregar los reportes FastQC de las lecturas hacia adelante y hacia detrás
En “Results “
“Which tool was used generate logs?: FastQC
En “FastQC output “ (Salida de FastQC)
“Type of FastQC output? “: Raw data
param-files“FastQC output “: archivos Raw data (salida de ambos FastQCtool)
Inspeccione la página web de MultiQC.
Preguntas
¿Qué opina de la calidad de las secuencias?
¿Qué debemos hacer?
La calidad de las secuencias parece peor para las lecturas inversas que para las lecturas hacia delante:
Puntuaciones de calidad por secuencia: distribución más a la izquierda, es decir, una menor calidad media de las secuencias
Calidad de la secuencia por base: curva menos suave y mayor disminución al final con un valor medio inferior al 28
Contenido de la secuencia por base: sesgo más fuerte al principio y sin distinción clara entre los grupos C-G y A-T
Los demás indicadores (adaptadores, niveles de duplicación, etc.) son similares.
Deberíamos recortar el final de las secuencias y filtrarlas con Cutadapttool
Con lecturas pareadas, las puntuaciones medias de calidad de las lecturas hacia delante serán casi siempre superiores a las de las lecturas hacia atrás.
Tras el recorte, las lecturas inversas serán más cortas debido a su calidad y se eliminarán durante el paso de filtrado. Si se elimina una de las lecturas inversas, debe eliminarse también su correspondiente lectura hacia adelante. De lo contrario, obtendremos un número diferente de lecturas en ambos archivos y en diferente orden, y el orden es importante para los siguientes pasos. Por lo tanto, es importante tratar las lecturas directas e inversas juntas para recortarlas y filtrarlas.
Práctica: Mejorando la calidad de los datos paired-end
Cutadapt ( Galaxy version 4.9+galaxy1) con los siguientes parámetros
“¿Lecturas de extremo único o de extremo pareado? “: Paired-end
No se encontraron adaptadores en estos conjuntos de datos. Cuando procese sus propios datos y sepa qué secuencias de adaptador se utilizaron durante la preparación de la biblioteca, debe proporcionar sus secuencias aquí.
En “Other Read Trimming Options “
“Quality cutoff(s) (R1) “: 20
En “Read Filtering Options “ (Opciones de filtrado de lectura)
“Minimum lenght (R1) “: 20
param-select“Resultados adicionales a generar “: Report
Inspeccione el archivo txt generado (Report)
Preguntas
¿Cuántos pares de bases se han eliminado de las lecturas debido a la mala calidad?
¿Cuántos pares de secuencias se han eliminado por ser demasiado cortos?
44.164bp (Quality-trimmed:) para las lecturas hacia adelante y 138.638bp para las lecturas hacia atrás.
Se han eliminado 1.376 secuencias porque al menos una lectura era más corta que el corte de longitud (322 cuando sólo se analizaron las lecturas hacia adelante).
Además del informe, Cutadapt genera 2 archivos:
Lectura 1 con las lecturas forward recortadas y filtradas
Lectura 2 con las lecturas inversas recortadas y filtradas
Estos conjuntos de datos pueden utilizarse para el análisis posterior, por ejemplo, el mapeo.
Preguntas
¿Qué tipo de alineación se utiliza para encontrar adaptadores en las lecturas?
¿Cuál es el criterio para elegir la mejor alineación de adaptadores?
Alineación semiglobal, es decir, sólo se utiliza para la puntuación la parte solapada de la lectura y la secuencia adaptadora.
Se calcula un alineamiento con solapamiento máximo que tiene el menor número de desajustes e indels.
Evaluar la calidad con Nanoplot - Sólo lecturas largas
En caso de lecturas largas, podemos comprobar la calidad de la secuencia con Nanoplot (De Coster et al. 2018). Proporciona estadísticas básicas con bonitos gráficos para un rápido control de calidad.
Práctica: Comprobación de la calidad de las lecturas largas
Crea un nuevo historial para esta parte y dale un nombre apropiado
Importe las lecturas PacBio HiFi m64011_190830_220126.Q20.subsample.fastq.gz de Zenodo
Figura 16: Comparación de Qscore entre Illumina, PacBio y Nanopore
Definición: Qscores es la probabilidad media de error por base, expresada en la escala log (Phred)
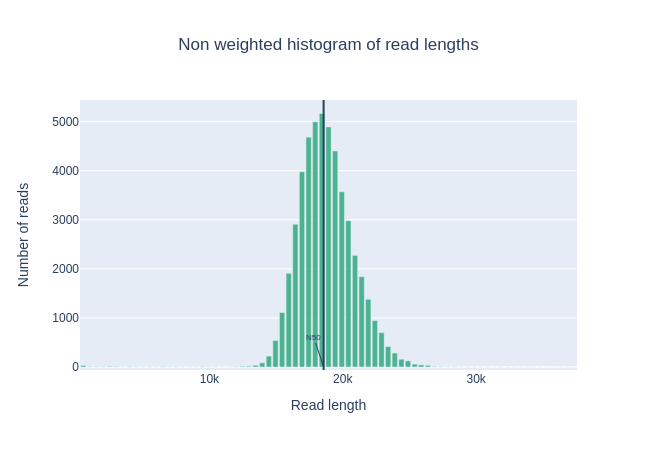
¿Cuál es la mediana, la media y N50?
La mediana, la longitud media de lectura y el N50 también están cerca de 18.000bp. Para las lecturas PacBio HiFi, la mayoría de las lecturas están generalmente cerca de este valor ya que la preparación de la biblioteca incluye un paso de selección de tamaño. Para otras tecnologías como PacBio CLR y Nanopore, es mayor y depende principalmente de la calidad de la extracción de ADN.
Histograma de longitudes de lectura
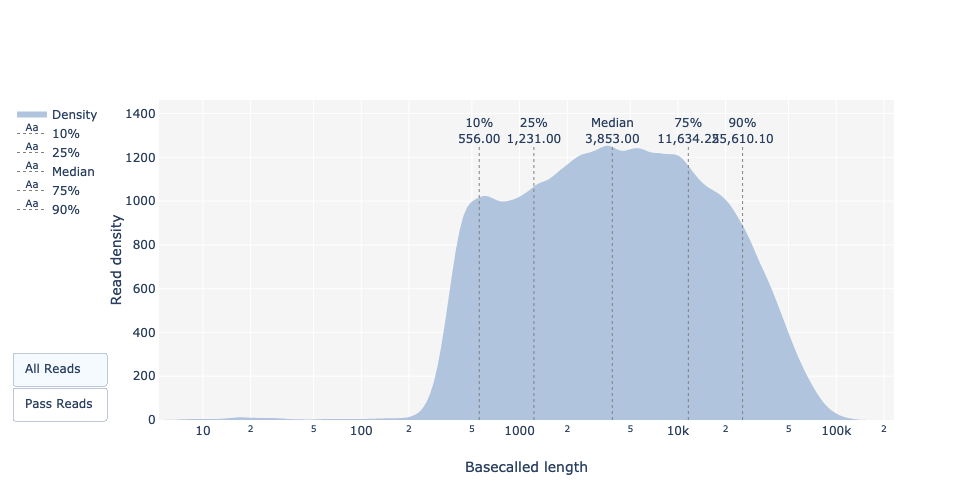
Este gráfico muestra la distribución de tamaños de fragmentos en el archivo analizado. A diferencia de la mayoría de las ejecuciones de Illumina, las lecturas largas tienen una longitud variable y esto mostrará las cantidades relativas de cada tamaño diferente de fragmento de secuencia. En este ejemplo, la distribución de la longitud de lectura se centra cerca de 18kbp pero los resultados pueden ser muy diferentes dependiendo de su experimento.
Gráfico de longitudes de lectura frente a calidad media de lectura mediante puntos
Este gráfico muestra la distribución del tamaño de los fragmentos en función de la puntuación Q del archivo analizado. En general, no hay relación entre la longitud de la lectura y la calidad de la lectura, pero esta representación permite visualizar ambas informaciones en un único gráfico y detectar posibles aberraciones. En runs con muchas lecturas cortas las lecturas más cortas son a veces de menor calidad que el resto.
Observando el “Gráfico de longitudes de lectura frente a calidad media de lectura utilizando el gráfico de puntos”. ¿Notaste algo inusual con el Qscore? ¿Puede explicarlo?
No hay lecturas bajo Q20. La calificación para las lecturas HiFi es:
Haz el control de calidad con FastQCtool en m64011_190830_220126.Q20.subsample.fastq.gz y compare los resultados!
Evaluar la calidad con PycoQC - Sólo Nanopore
PycoQC (Leger and Leonardi 2019) es una herramienta de visualización de datos y control de calidad para datos de nanoporos. A diferencia de FastQC/Nanoplot, necesita un archivo específico sequencing_summary.txt generado por Oxford nanopore basecallers como Guppy o el antiguo albacore basecaller.
Uno de los puntos fuertes de PycoQC es que es interactivo y altamente personalizable, por ejemplo, los gráficos se pueden recortar, se puede acercar y alejar, sub-seleccionar áreas y exportar figuras.
Práctica: Comprobación de calidad de las lecturas Nanopore
Crea un nuevo historial para esta parte y dale un nombre apropiado
Importar las lecturas nanopore nanopore_basecalled-guppy.fastq.gz y sequencing_summary.txt de Zenodo
~270k lecturas en total (ver la tabla de resumen de Basecall, “All reads”) Para la mayoría de los perfiles de basecall, Guppy asignará lecturas como “Pass” si el Qscore de la lectura es al menos igual a 7.
¿Cuál es la mediana, el mínimo y el máximo de longitud de lectura, y cuál es el N50?
La mediana de la longitud de lectura y el N50 se pueden encontrar para todos, así como para todas las lecturas aprobadas, es decir, las lecturas que pasaron los ajustes de calidad de Guppy (Qscore >= 7), en la tabla de resumen basecall. Para las longitudes de lectura mínima (195bp) y máxima (256kbp), se puede encontrar con el gráfico de longitudes de lectura.
Longitud de las lecturas base
Como para FastQC y Nanoplot, este gráfico muestra la distribución de tamaños de fragmentos en el archivo analizado. En cuanto a PacBio CLR/HiFi, las lecturas largas tienen una longitud variable y esto mostrará las cantidades relativas de cada tamaño diferente de fragmento de secuencia. En este ejemplo, la distribución de la longitud de lectura es bastante dispersa, con una longitud de lectura mínima para las lecturas pasadas de alrededor de 200bp y una longitud máxima de ~150.000bp.
Este gráfico muestra la distribución de las puntuaciones de calidad (Q) para cada lectura. Esta puntuación pretende dar una puntuación de calidad global para cada lectura. La definición exacta de Qscores es: la probabilidad media de error por base, expresada en la escala log (Phred). En el caso de los datos Nanopore, la distribución se centra generalmente en torno a 10 o 12. En el caso de ejecuciones antiguas, la distribución puede ser menor, ya que los modelos de basecalling son menos precisos que los modelos recientes.
Longitud de las lecturas por base frente a la calidad de las lecturas PHRED
Preguntas
¿Qué aspecto tienen la calidad media y la distribución de la calidad del experimento?
La mayoría de las lecturas tienen un Qscore entre 8 y 11 que es estándar para los datos Nanopore. Tenga en cuenta que para los mismos datos, el basecaller utilizado (Albacor, Guppy, Bonito), el modelo (fast, hac, sup) y la versión de la herramienta pueden dar resultados diferentes.
Al igual que NanoPlot, esta representación ofrece una visualización en 2D de la puntuación Q de las lecturas en función de su longitud.
Figura 22: Basecalled reads length vs reads PHRED quality
Resultados durante el experimento
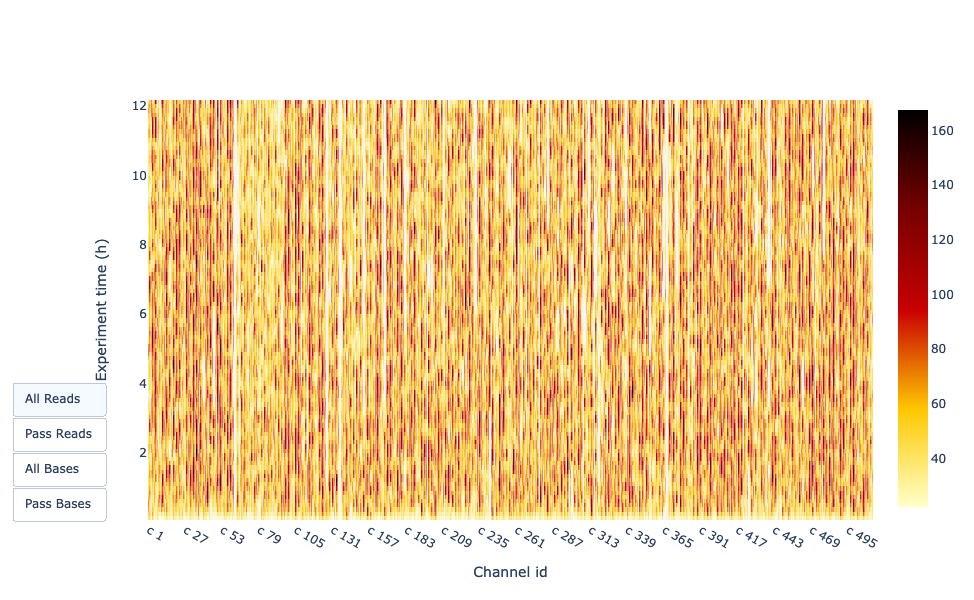
Esta representación ofrece información sobre las lecturas secuenciadas a lo largo del tiempo para una única ejecución:
Cada imagen indica una nueva carga de la celda de flujo (3 + la primera carga).
La contribución en lecturas totales para cada “recarga”.
La producción de lecturas está disminuyendo con el tiempo:
Se secuencia la mayor parte del material (ADN/ARN)
Saturación de poros
Degradación de material/poros
…
En este ejemplo, la contribución de cada repostaje es muy baja, y puede considerarse una mala ejecución. El área del gráfico “Acumulativo” (azul claro) indica que el 50% de todas las lecturas y casi el 50% de todas las bases se produjeron en las primeras 5h del experimento de 25h. Aunque es normal que el rendimiento disminuya con el tiempo, una disminución como ésta no es una buena señal.
En este ejemplo, la producción de datos a lo largo del tiempo sólo disminuyó ligeramente durante las 12h con un aumento continuo de los datos acumulados. La ausencia de una curva decreciente al final del experimento indica que todavía hay material biológico en la celda de flujo. El experimento finalizó antes de que se secuenciara todo. Es una corrida excelente, incluso puede considerarse como excepcional.
Longitud de lectura durante el experimento
Preguntas
¿Ha cambiado la longitud de la lectura con el tiempo? ¿Cuál podría ser la razón?
En el ejemplo actual, la longitud de la lectura aumenta con el tiempo de la carrera de secuenciación. Una explicación es que la densidad del adaptador es mayor para muchos fragmentos cortos y, por tanto, la posibilidad de que un fragmento más corto se adhiera a un poro es mayor. Además, las moléculas más cortas pueden desplazarse más rápidamente por el chip. Sin embargo, con el tiempo, los fragmentos más cortos son cada vez más raros y, por tanto, son más los fragmentos largos que se adhieren a los poros y se secuencian.
La longitud de lectura a lo largo del experimento debería ser estable. Puede aumentar ligeramente a lo largo del tiempo, ya que los fragmentos cortos tienden a sobre-secuenciarse al principio y están menos presentes a lo largo del tiempo.
Figura 24: Longitud de lectura sobre tiempo de experimento
Actividad del canal en el tiempo
Ofrece una visión general de los poros disponibles, el uso de los poros durante el experimento, los poros inactivos y muestra si la carga de la celda de flujo es buena (se utilizan casi todos los poros). En este caso, la gran mayoría de canales/poros están inactivos (blanco) durante todo el experimento de secuenciación, por lo que el experimento puede considerarse malo.
Es de esperar que el gráfico sea oscuro cerca del eje X y que con valores Y más altos (tiempo creciente) no se vuelva demasiado claro/blanco. Dependiendo de si elige “Reads” o “Bases” a la izquierda, el color indica el número de bases o de lecturas por intervalo de tiempo
En este ejemplo, casi todos los poros están activos a lo largo de la secuencia (perfil amarillo/rojo), lo que indica una secuencia excelente.
Comentario: ¡Pruébelo!
Haga el control de calidad con FastQCtool y/o Nanoplottool en nanopore_basecalled-guppy.fastq.gz y compare los resultados!
Conclusión
En este tutorial comprobamos la calidad de los archivos FASTQ para asegurarnos de que sus datos tienen buen aspecto antes de inferir más información. Este paso es el primer paso habitual para análisis como RNA-Seq, ChIP-Seq, o cualquier otro análisis OMIC que dependa de datos NGS. Los pasos de control de calidad son similares para cualquier tipo de datos de secuenciación:
Evaluación de la calidad con herramientas como:
Lecturas cortas: FASTQE ( Galaxy version 0.3.1+galaxy0)
Corto+Largo: FASTQC ( Galaxy version 0.73+galaxy0)
Lecturas largas: Nanoplot ( Galaxy version 1.41.0+galaxy0)
Sólo Nanopore: PycoQC ( Galaxy version 2.5.2+galaxy0)
Recorte y filtrado de lecturas cortas con una herramienta como Cutadapttool
You've finished the tutorial
Please also consider filling out the Feedback Form as well!
Puntos clave
Realiza controles de calidad en cada conjunto de datos antes de ejecutar cualquier otro análisis bioinformático.
Evalúa las métricas de calidad y mejora la calidad si es necesario.
Comprueba el impacto del control de calidad.
Están disponibles diferentes herramientas para proporcionar métricas de calidad adicionales.
Para lecturas paired‑end, analiza juntas las lecturas forward y reverse.
Preguntas frecuentes
Have questions about this tutorial? Have a look at the available FAQ pages and support channels
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Ewels, P., M. Magnusson, S. Lundin, and M. K\~ A\textcurrencyller, 2016 MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048. 10.1093/bioinformatics/btw354
De Coster, W., S. D’Hert, D. T. Schultz, M. Cruts, and C. Van Broeckhoven, 2018 NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34: 2666–2669. 10.1093/bioinformatics/bty149
Leger, A., and T. Leonardi, 2019 pycoQC, interactive quality control for Oxford Nanopore Sequencing . Journal of Open Source Software 4: 1236. 10.21105/joss.01236
Jacques, R. M. S., W. M. Maza, S. D. Robertson, A. Lonsdale, C. S. Murray et al., 2021 A Fun Introductory Command Line Lesson: Next Generation Sequencing Quality Analysis with Emoji! CourseSource 8: 10.24918/cs.2021.17
Retroalimentación
¿Utilizaste este material como instructor? Cuéntanos tu experiencia.
¿Has usado este material como aprendiz o estudiante? Haz click en el formulario a continuación para dejarnos tu opinión
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{sequence-analysis-quality-control,
author = "Bérénice Batut and Maria Doyle and Alexandre Cormier and Anthony Bretaudeau and Laura Leroi and Erwan Corre and Stéphanie Robin and Cameron Hyde",
title = "Control de calidad (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/sequence-analysis/tutorials/quality-control/tutorial_ES.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Referencias
These individuals or organisations provided funding support for the development of this resource
Preguntas:

 Open image in new tab
Open image in new tab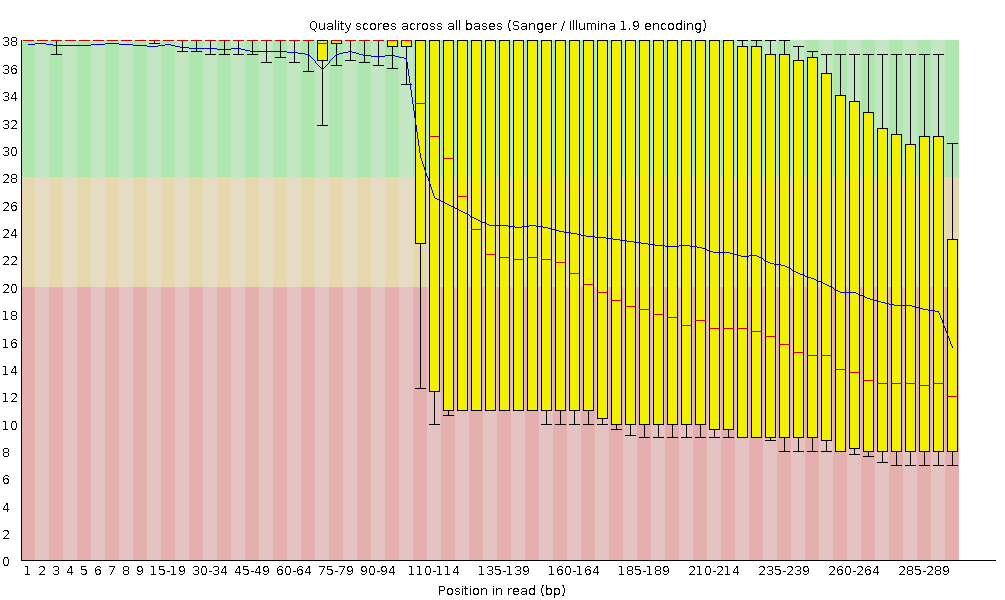 Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab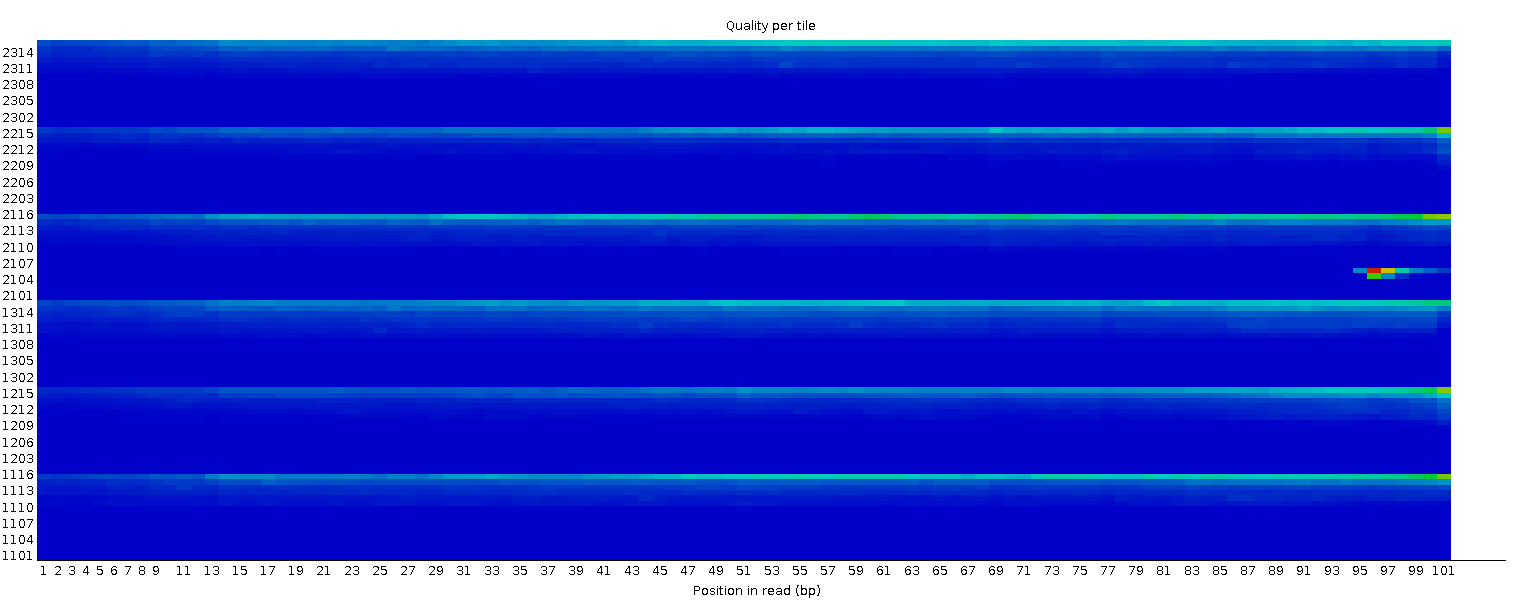
 Open image in new tab
Open image in new tab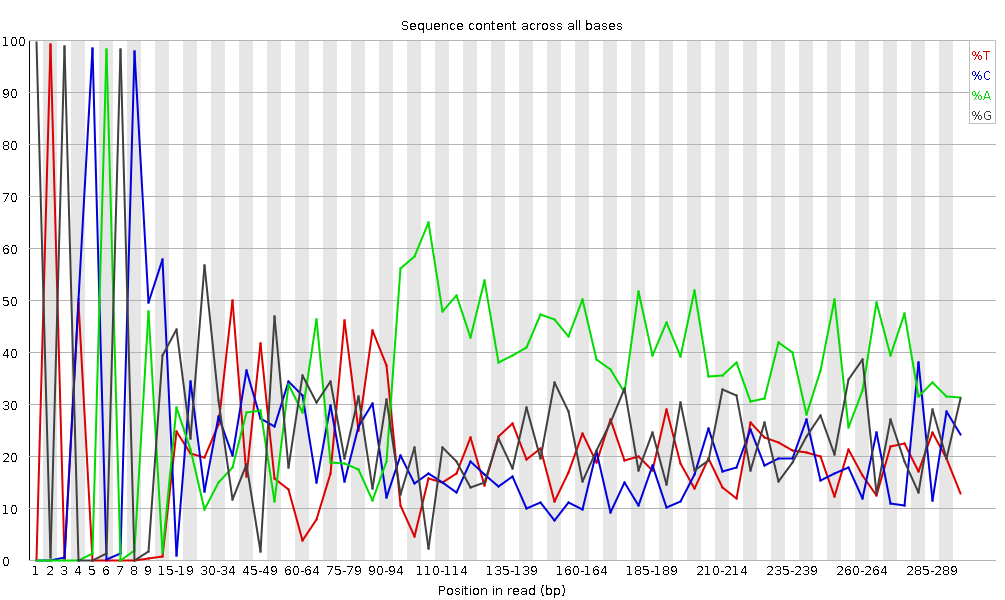 Open image in new tab
Open image in new tab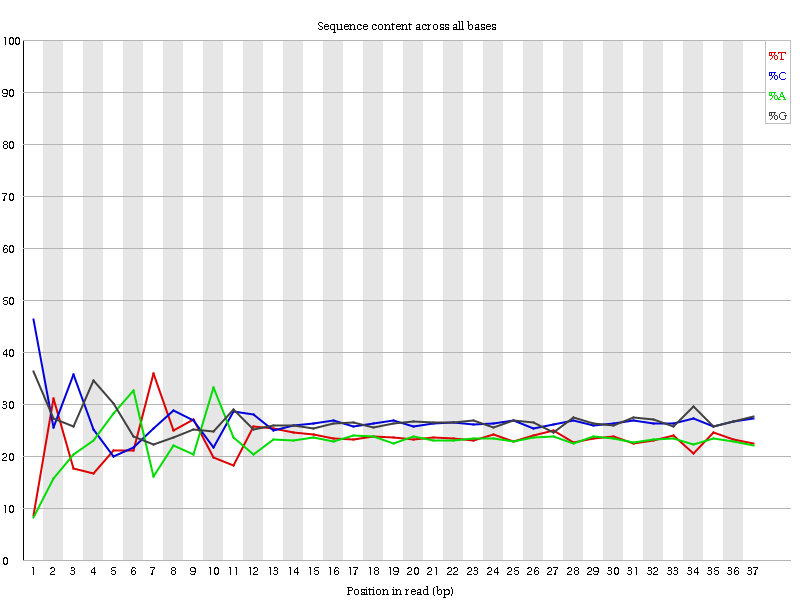
 Open image in new tab
Open image in new tab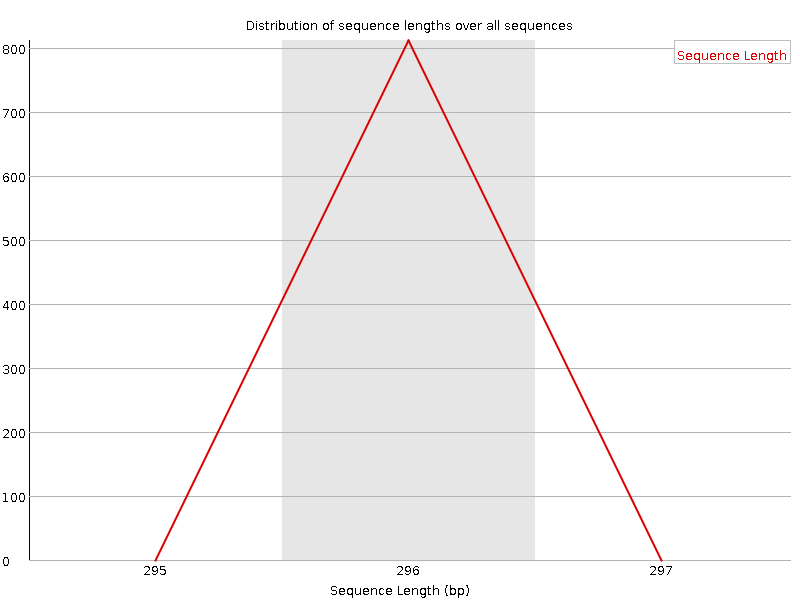 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab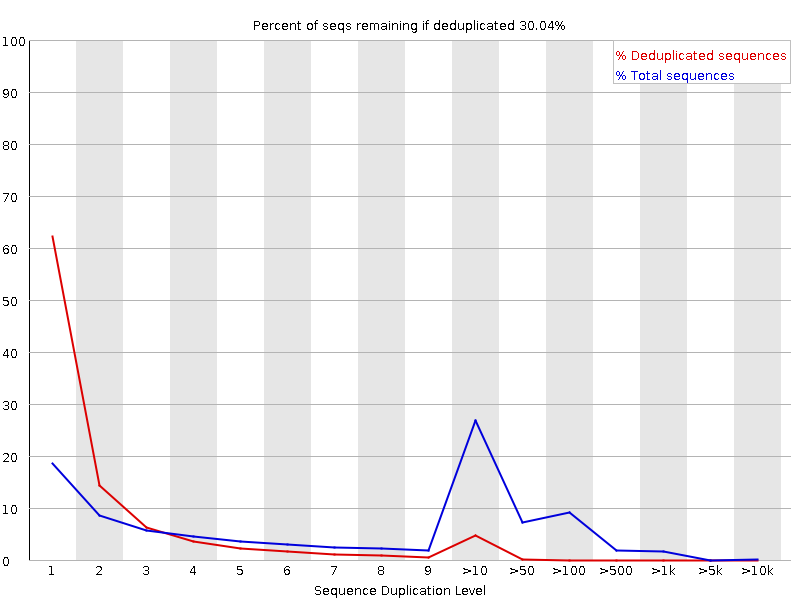
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab

Tenemos algunas rayas rojas porque hemos recortado esas regiones de las lecturas.
Ahora tenemos un pico de alta calidad en lugar de uno de alta y otro de baja calidad que teníamos anteriormente.
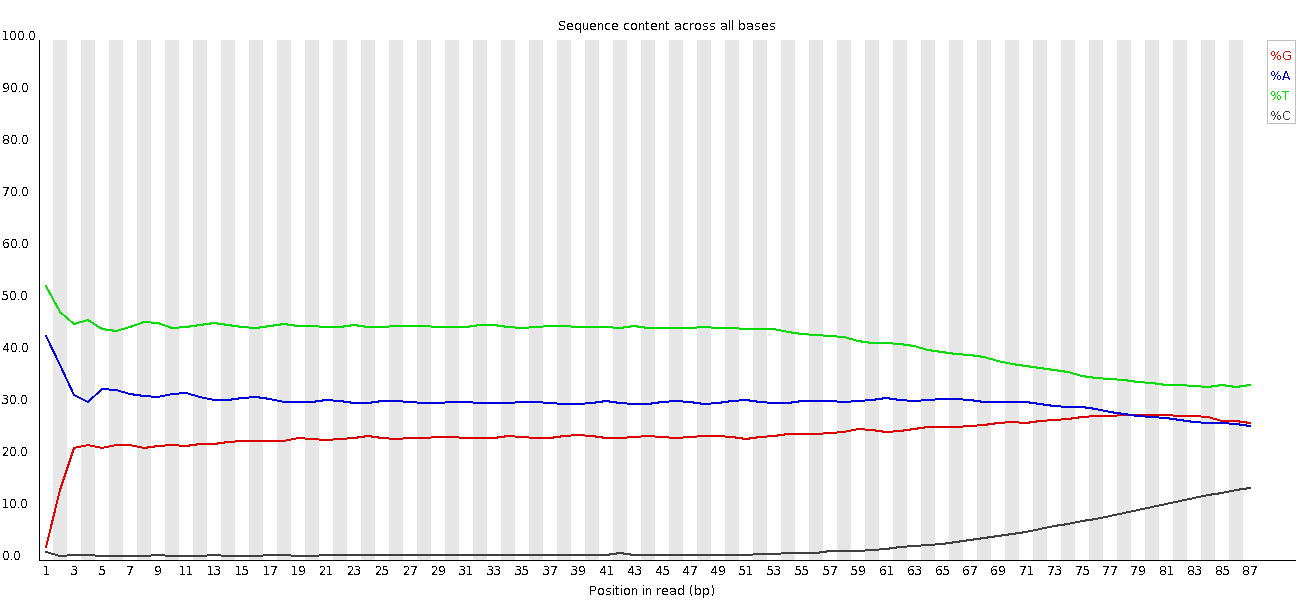
No tenemos la misma representación de las bases que antes, ya que se trata de datos de amplicones.
Ahora tenemos un único pico principal de GC debido a la eliminación del adaptador.
Esto es lo mismo que antes, ya que no tenemos Ns en estas lecturas.
Ahora tenemos múltiples picos y un rango de longitudes, en lugar del único pico que teníamos antes del recorte cuando todas las secuencias tenían la misma longitud.
Open image in new tab
 Open image in new tab
Open image in new tab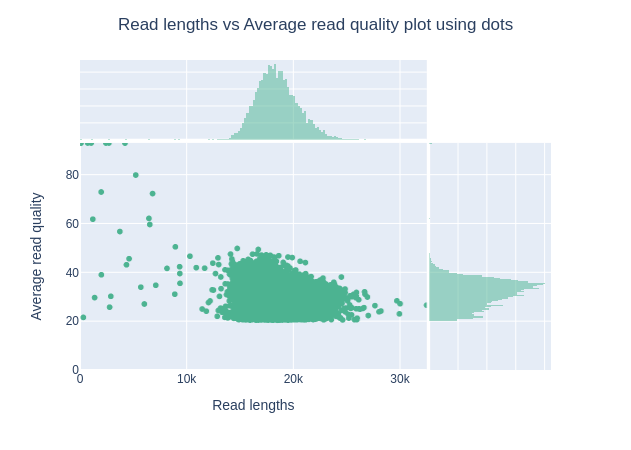 Open image in new tab
Open image in new tabOpen image in new tab
 Open image in new tab
Open image in new tab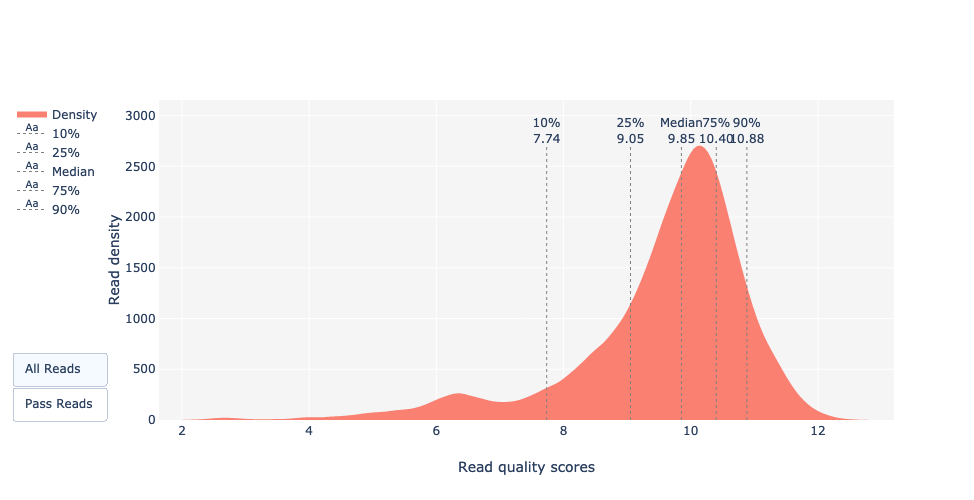 Open image in new tab
Open image in new tab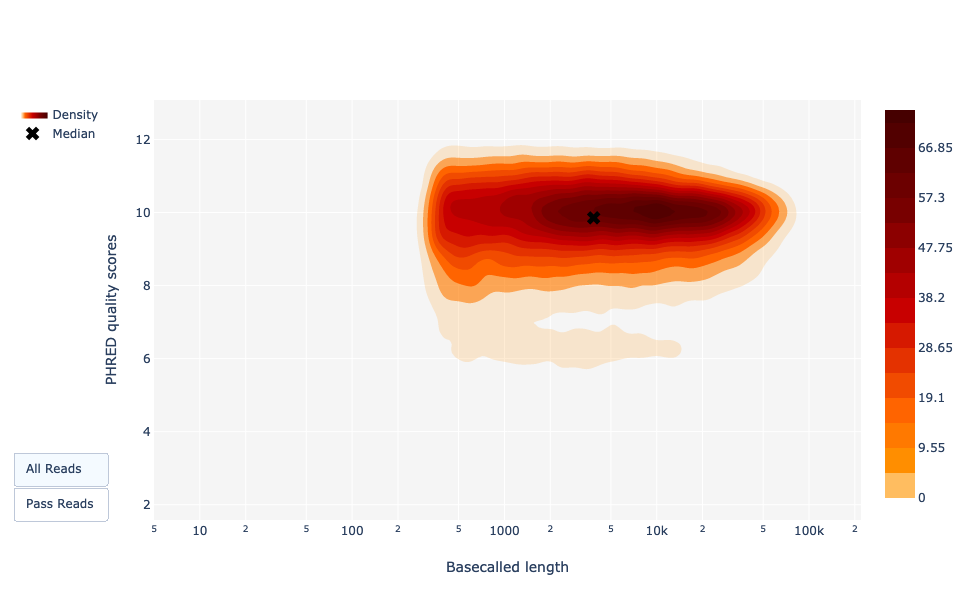 Open image in new tab
Open image in new tab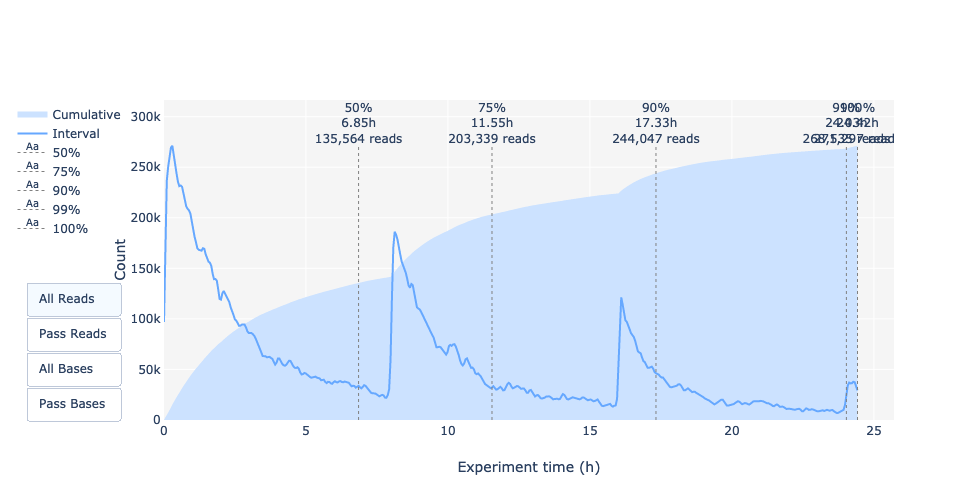 Open image in new tab
Open image in new tab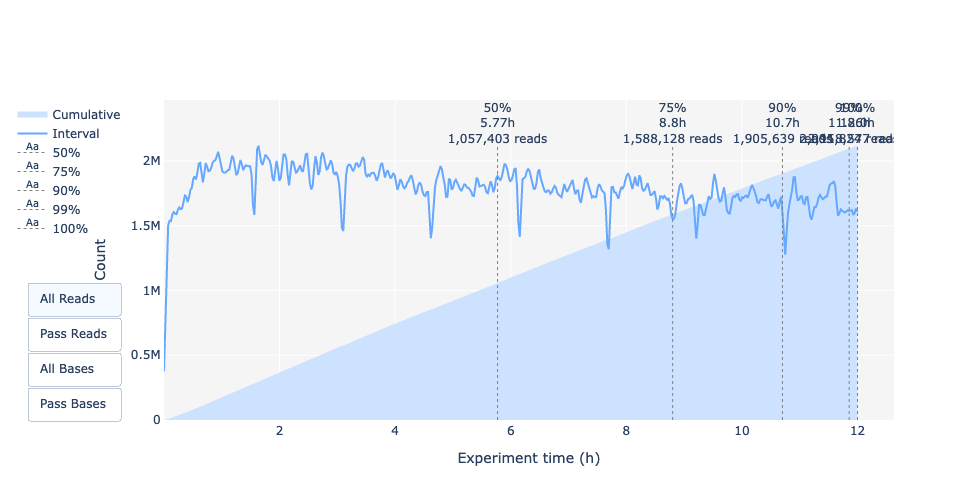
 Open image in new tab
Open image in new tab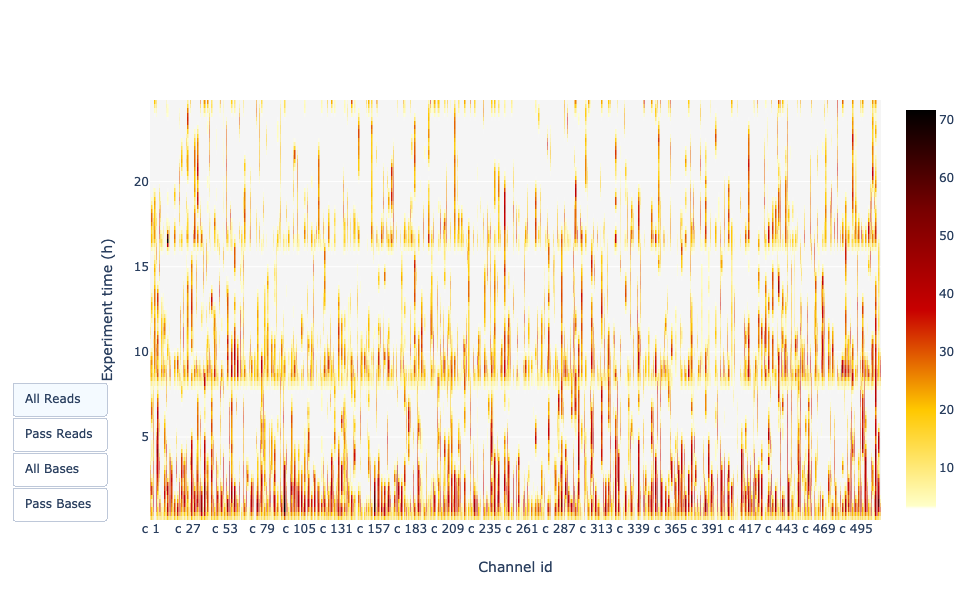 Open image in new tab
Open image in new tab