En los últimos años, la secuenciación del ARN (abreviada RNA-Seq) se ha convertido en una tecnología muy utilizada para analizar el transcriptoma celular en continuo cambio, es decir, el conjunto de todas las moléculas de ARN de una célula o de una población de células. Uno de los objetivos más comunes de RNA-Seq es el perfilado de la expresión génica mediante la identificación de genes o rutas moleculares que se expresan de forma diferencial (DE) entre dos o más condiciones biológicas. Este tutorial muestra un flujo de trabajo computacional para la detección de genes y rutas de expresión diferencial a partir de datos de ARN-Seq, proporcionando un análisis completo de un experimento de ARN-Seq en células de Drosophila tras la eliminación de un gen regulador.
En el estudio de Brooks et al. 2011, los autores identificaron genes y vías reguladas por el gen Pasilla (el homólogo en Drosophila de las proteínas reguladoras del splicing en mamíferos Nova-1 y Nova-2) usando datos de RNA-Seq. Redujeron el gen Pasilla (PS) en Drosophila melanogaster mediante ARN de interferencia (ARNi). A continuación, se aisló el ARN total y se utilizó para preparar bibliotecas de ARN-Seq de extremo único y de extremo pareado para muestras tratadas (sin PS) y no tratadas. Estas bibliotecas se secuenciaron para obtener las lecturas de RNA-Seq de cada muestra. Los datos de RNA-Seq de las muestras tratadas y no tratadas pueden compararse para identificar los efectos de la depleción del gen Pasilla en la expresión génica.
En este tutorial, ilustramos paso a paso el análisis de los datos de expresión génica utilizando 7 de los conjuntos de datos originales:
Cada muestra constituye una réplica biológica separada de la condición correspondiente (tratada o no tratada). Además, dos de las muestras tratadas y dos de las no tratadas proceden de un ensayo de secuenciación de extremo pareado, mientras que las muestras restantes proceden de un experimento de secuenciación de extremo único.
Comentario: Datos completos
Los datos originales están disponibles en NCBI Gene Expression Omnibus (GEO) con el número de acceso GSE18508. Las lecturas de ARN-Seq en bruto se han extraído de los archivos Sequence Read Archive (SRA) y se han convertido en archivos FASTQ.
En la primera parte de este tutorial utilizaremos los archivos de 2 de las 7 muestras para demostrar cómo calcular el recuento de lecturas (una medida de la expresión génica) a partir de archivos FASTQ (control de calidad, mapeo, recuento de lecturas). Proporcionamos los archivos FASTQ para las otras 5 muestras por si desea reproducir todo el análisis más adelante.
En la segunda parte del tutorial, se utilizan los recuentos de lecturas de las 7 muestras para identificar y visualizar los genes, familias de genes y rutas moleculares DE debido a la depleción del gen PS.
Práctica: Carga de datos
Crear un nuevo historial para este ejercicio de RNA-Seq
Haz click sobre el icono new-history en la parte superior del panel de historiales.
Importar los pares de archivos FASTQ desde Zenodo o una biblioteca de datos:
Abre el manejador de carga de datos de Galaxy (galaxy-upload (Upload) en la parte superior derecha del panel de herramientas)
Selecciona ‘Pegar/Traer datos’ Paste/Fetch Data
Copia los enlaces en el campo de textos
Presiona ‘Iniciar’ Start
Close Cierra la ventana.
Galaxy utiliza los URLs como nombres de forma predeterminada , así que los tendrás que cambiar a algunos que sean más útiles o informativos.
Como alternativa a cargar los datos desde una URL o desde su ordenador, los archivos también pueden estar disponibles desde una biblioteca de datos compartidos:
Entra en Libraries (panel izquierdo)
Navega a la carpeta correcta indicada por su instructor. - En la mayoría de los tutoriales de Galaxias los datos serán proporcionados en una carpeta llamada GTN - Material –> Topic name -> Tutorial name.
Seleccione los archivos deseados
Haz clic en Add to Historygalaxy-dropdown cerca de la parte superior y selecciona as Datasets en el menú desplegable
En la ventana emergente, elige
“Seleccionar historial “: el historial al que desea importar los datos (o crear uno nuevo)
Haga clic en Import
Comentario
Tenga en cuenta que estos son los archivos completos de las muestras y ~1.5Gb cada uno, por lo que puede tardar algunos minutos en importarse.
Para una rápida ejecución de los pasos FASTQ un pequeño subconjunto de cada archivo FASTQ (~ 5Mb) se puede encontrar aquí en Zenodo:
Compruebe que el tipo de datos es fastqsanger (por ejemplo, nofastq). Si no lo es, cambie el tipo de datos a fastqsanger.
Selecciona sobre el galaxy-pencilicono del lápiz para editar los atributos del conjunto de datos
Selecciona en la pestaña galaxy-chart-select-dataDatatypes en la parte superior del panel central
Selecciona fastqsanger
Da clic en el botón Change datatype
Cree una colección emparejada llamada 2 PE fastqs, nombre sus pares con el nombre de la muestra seguido de los atributos: GSM461177_untreat_paired y GSM461180_treat_paired.
Haga clic en galaxy-selectorSelect Items en la parte superior del panel del historial
Marque todos los conjuntos de datos de su historial que desee incluir
Haga clic en n of N selected y elija Build List of Dataset Pairs
Cambie el texto de unpaired forward por un selector común para las lecturas forward
Cambie el texto de unpaired reverse por un selector común para las lecturas inversas
Haga clic en Pair this dataset para cada par válido anterior e inverso.
Introduzca un nombre para su colección
Haga clic en Create list para crear su colección
Vuelva a hacer clic en el icono de marca de verificación situado en la parte superior de su historial
Preguntas
¿Cómo se guardan las secuencias de ADN?
¿Qué son las otras entradas del fichero?
Las secuencias de ADN se guardan en un archivo FASTQ, en la segunda línea de cada grupo de cuatro línesa.
Este formato de archivo es llamado FASTQ. Almacena información de secuenciamientos y de calidad. Cada secuancia está representada por un grupo de 4 líneas con la primera siendo la primera el identificador de la secuencia, la segunda la secuencia de nucleótidos, la tercera es una línea de transición, y la última una secuencia de la puntuación de calidad para cada nucleótido. each nucleotide.
Las lecturas son datos brutos de la máquina de secuenciación sin ningún tratamiento previo. Es necesario evaluar su calidad.
Control de calidad
Durante la secuenciación se introducen errores, como la llamada de nucleótidos incorrectos. Esto se debe a las limitaciones técnicas de cada plataforma de secuenciación. Los errores de secuenciación pueden sesgar el análisis y dar lugar a una interpretación errónea de los datos. También puede haber adaptadores si las lecturas son más largas que los fragmentos secuenciados, y recortarlos puede mejorar el número de lecturas mapeadas.
El control de calidad de la secuencia es, por tanto, un primer paso esencial en su análisis. Utilizaremos herramientas similares a las descritas en el tutorial “Control de calidad”:
Falco, que es una reescritura optimizada para la eficiencia de FastQC, para crear un informe de la calidad de la secuencia
Cutadapt (Marcel 2011) para mejorar la calidad de las secuencias mediante el recorte y el filtrado.
Lamentablemente, la versión actual de MultiQC (la herramienta que utilizamos para combinar informes) no admite colecciones de listas de pares. Primero tendremos que transformar nuestra lista de pares en una lista simple.
La situación actual está en la parte superior y la herramienta Aplanar colección la transformará en la situación que se muestra en la parte inferior:
Flatten collection con los siguientes parámetros convertir la lista de pares en una lista simple:
“Input Collection “: 2 PE fastqs
Falco ( Galaxy version 1.2.4+galaxy0) con los siguientes parámetros:
param-collection“Raw read data from your current history “: Salida de Flatten collectiontool seleccionada como Dataset collection
Haga clic en param-collectionDataset collection delante del parámetro de entrada al que desea suministrar la colección.
Seleccione la colección que desea utilizar de la lista
Inspeccionar la página web de salida de Falcotool para la muestra GSM461177_untreat_paired (forward y reverse)
Preguntas
¿Cuál es la longitud de la lectura?
La longitud de lectura de ambas parejas es de 37bp.
Como es tedioso inspeccionar todos estos informes individualmente los combinaremos con MultiQC ( Galaxy version 1.11+galaxy1).
MultiQC ( Galaxy version 1.24.1+galaxy0) para agregar los informes Falco con los siguientes parámetros:
En “Results “:
“Results “
“Which tool was used generate logs?: FastQC (Falco es un sustituto directo de FastQC y podemos pasar su salida a MultiQC como si hubiera sido generada por la herramienta original.)
En “FastQC output”:
param-repeat“Insert FastQC output”
param-collection“FastQC output”: Falco on collection N: RawData (salida de Falcotool)
Inspeccionar la página web de salida de MultiQC para cada FASTQ
Preguntas
¿Qué opina de la calidad de las secuencias?
¿Qué debemos hacer?
Todo parece correcto para 3 de los archivos. El GSM461177_untreat_paired tiene 10,6 millones de secuencias emparejadas y el GSM461180_treat_paired 12,3 millones de secuencias emparejadas. Pero en GSM461180_treat_paired_reverse (reverse reads de GSM461180) la calidad disminuye bastante al final de las secuencias.
Todos los archivos excepto GSM461180_treat_paired_reverse tienen una alta proporción de lecturas duplicadas (esperable en datos RNA-Seq).
La “calidad de la secuencia por base” es globalmente buena, con un ligero descenso al final de las secuencias. Para GSM461180_treat_paired_reverse, la disminución es bastante grande.
Casi no hay adaptadores conocidos ni secuencias sobrerrepresentadas.
Si la calidad de las lecturas es mala, deberíamos:
Compruebe qué está mal y piense en las posibles razones de la mala calidad de las lecturas: puede deberse al tipo de secuenciación o a lo que hemos secuenciado (gran cantidad de secuencias sobrerrepresentadas en datos transcriptómicos, porcentaje sesgado de bases en datos Hi-C)
Pregunte al centro de secuenciación al respecto
Realizar algún tratamiento de calidad (teniendo cuidado de no perder demasiada información) con algún recorte o eliminación de malas lecturas
Debemos recortar las lecturas para eliminar las bases que se secuenciaron con alta incertidumbre (es decir, bases de baja calidad) en los extremos de las lecturas, y también eliminar las lecturas de mala calidad general.
Preguntas
¿Cuál es la relación entre GSM461177_untreat_paired_forward y GSM461177_untreat_paired_reverse ?
Los datos han sido secuenciados utilizando secuenciación paired-end (de extremos emparejados).
La secuenciación paired-end se basa en la idea de que los fragmentos iniciales de ADN (más largos que la longitud de lectura real) se secuencian desde ambos extremos. Este enfoque da lugar a dos lecturas por fragmento: la primera en orientación directa y la segunda en orientación inversa complementaria. La distancia entre ambas lecturas es conocida, por lo que puede utilizarse como información adicional para mejorar el mapeo de las lecturas.
Con la secuenciación paired-end, cada fragmento queda más cubierto que con la secuenciación single-end (solo se secuencia en orientación directa):
La secuenciación paired-end genera entonces dos ficheros:
Un fichero con la secuenciación correspondiente a la orientación hacia adelante de todos los fragmentos
Un fichero con la secuenciación correspondiente a la orientación inversa de todos los fragmentos
Aquí, GSM461177_untreat_paired_forward corresponde a las lecturas hacia adelante y GSM461177_untreat_paired_reverse a las inversas.
Práctica: Recorte de FASTQs
Cutadapt ( Galaxy version 4.9+galaxy1) con los siguientes parámetros para recortar las secuencias de baja calidad:
“Single-end or Paired-end reads?: Paired-end Collection
param-collection“Paired Collection “: 2 PE fastqs
En “Other Read Trimming Options”
“Quality cutoff(s) (R1)”: 20
En “Read Filtering Options”
“Minimum length (R1)”: 20
En “Additional outputs to generate”
Seleccionar: Report: Cutadapt's per-adapter statistics. You can use this file with MultiQC.
Preguntas
Por qué ejecutamos la herramienta de recorte solo una vez en un conjunto de datos paired-end y no dos veces, una para cada conjunto de datos?
La herramienta puede eliminar secuencias si se vuelven demasiado cortas durante el proceso de recorte. En archivos paired-end, elimina pares de secuencias completos si una (o ambas) de las dos lecturas quedan por debajo del umbral de longitud establecido. Las lecturas de un par que superan dicho umbral, pero cuya pareja ha quedado demasiado corta, pueden opcionalmente guardarse en archivos single-end. Esto garantiza que no se pierda por completo la información de un par de lecturas si solo una de ellas es de buena calidad.
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los informes Cutadapt con los siguientes parámetros:
En “Results”:
param-repeat“Results”
“Which tool was used generate logs?”: Cutadapt/Trim Galore!
param-collection“Output of Cutadapt”: Cutadapt on collection N: Report (salida de Cutadapttool) seleccionada como Dataset collection
Preguntas
¿Cuántos pares de secuencias se han eliminado porque al menos una lectura era más corta que el corte de longitud?
¿Cuántos pares de bases se han eliminado de las lecturas directas debido a la mala calidad? ¿Y de las lecturas inversas?
147.810 (1,4%) lecturas eran demasiado cortas para GSM461177_untreat_paired y 1.101.875 (9%) para GSM461180_treat_paired.
La salida MultiQC sólo proporciona la proporción de pb recortados en total, no para cada lectura. Para obtener esta información, es necesario volver a los informes individuales. Para GSM461177_untreat_paired, se han recortado 5.072.810 pb de las lecturas directas (Lectura 1) y 8.648.619 pb de las lecturas inversas (Lectura 2) debido a la calidad. En el caso de GSM461180_treat_paired, se han recortado 10.224.537 pb de las lecturas anteriores y 51.746.850 pb de las inversas. Esto no es una sorpresa; vimos que al final de las lecturas la calidad disminuía más para las lecturas inversas que para las lecturas delanteras, especialmente para GSM461180_treat_paired.
Mapeo
Para dar sentido a las lecturas, primero tenemos que averiguar de dónde proceden las secuencias en el genoma, para poder determinar a qué genes pertenecen. Cuando se dispone de un genoma de referencia del organismo, este proceso se conoce como alineación o “mapeo” de las lecturas con la referencia. Esto equivale a resolver un rompecabezas, pero, por desgracia, no todas las piezas son únicas.
Comentario
¿Desea obtener más información sobre los principios en los que se basa el mapeo? Siga nuestra formación.
En este estudio, los autores utilizaron células de Drosophila melanogaster. Por lo tanto, debemos mapear las secuencias de calidad controlada al genoma de referencia de Drosophila melanogaster.
Preguntas
¿Qué es un genoma de referencia?
Por cada organismo modelo, muchos genomas de referencia pueden estar disponibles (p.e. hg19 y hg38 para humanos). ¿A qué corresponden?
¿Qué genoma de referencia deberíamos usar?
Un genoma de referencia (o ensamblado de referencia) es un conjunto de secuencias de ácidos nucleicos ensambladas como un ejemplo representativo del material genético de una especie. Dado que suelen ensamblarse a partir de la secuenciación de diferentes individuos, no representan con exactitud el conjunto de genes de un solo organismo, sino un mosaico de distintas secuencias de ácidos nucleicos provenientes de cada individuo.
A medida que disminuye el costo de la secuenciación del ADN y surgen nuevas tecnologías de secuenciación de genomas completos, se generan cada vez más secuencias genómicas. Con estas nuevas secuencias, se construyen nuevas alineaciones y se mejoran los genomas de referencia (con menos huecos, corrección de errores en la secuencia, etc.). Los distintos genomas de referencia corresponden a las diferentes versiones publicadas, conocidas como “ensamblados” o builds.
El genoma de Drosophila melanogaster es conocido y ensamblado y puede utilizarse como genoma de referencia en este análisis. Tenga en cuenta que pueden publicarse nuevas versiones de genomas de referencia si el ensamblaje mejora, para este tutorial vamos a utilizar la versión 6 del ensamblaje del genoma de referencia de Drosophila melanogaster(dm6).
En los transcriptomas de eucariotas, la mayoría de las lecturas proceden de ARNm procesados que carecen de intrones:
Figura 9: Los tipos de lecturas RNA-seq (adaptación de la Figura 1a de Kim et al. 2015): lecturas que mapean completamente dentro de un exón (en rojo), lecturas que abarcan más de 2 exones (en azul), lecturas que abarcan más de 2 exones (en púrpura)
Por lo tanto no pueden ser simplemente mapeadas de vuelta al genoma como hacemos normalmente con los datos de ADN. Se han desarrollado mapeadores empalmados para mapear eficientemente las lecturas derivadas de transcripciones contra un genoma de referencia:
Figura 10: Principio de los mapeadores empalmados: (1) identificación de las lecturas que abarcan un único exón, (2) identificación de las uniones de empalme en las lecturas no mapeadas
En los últimos años se han desarrollado varios mapeadores empalmados para procesar la explosión de datos de RNA-Seq.
TopHat (Trapnell et al. 2009) fue una de las primeras herramientas diseñadas específicamente para abordar este problema. En TopHat las lecturas se mapean contra el genoma y se separan en dos categorías: (1) las que se mapean, y (2) las que están inicialmente sin mapear (IUM). las “pilas” de lecturas que representan posibles exones se extienden en busca de posibles sitios de empalme donante/aceptor y se reconstruyen las posibles uniones de empalme. A continuación, se mapean los IUM en estas uniones.
Para optimizar y acelerar aún más la alineación de lecturas empalmadas, se desarrolló HISAT2 (Kim et al. 2019). Utiliza un índice de grafo jerárquico FM (HGFM), que representa el genoma completo y las posibles variantes, junto con índices locales superpuestos (cada uno de los cuales abarca ~57 kb) que cubren colectivamente el genoma y sus variantes. Esto permite encontrar ubicaciones semilla iniciales para potenciales alineaciones de lecturas en el genoma utilizando el índice global y refinar rápidamente estas alineaciones utilizando un índice local correspondiente:
Figura 13: Índice FM del gráfico jerárquico en HISAT/HISAT2 (Figura S8 de Kim et al. 2015)
Una parte de la lectura (flecha azul) se mapea primero en el genoma utilizando el índice FM global. HISAT2 intenta entonces extender el alineamiento directamente utilizando la secuencia del genoma (flecha violeta). En (a) tiene éxito y esta lectura se alinea ya que reside completamente dentro de un exón. En (b) la extensión encuentra un desajuste. Ahora HISAT2 se aprovecha del índice FM local que se solapa con esta localización para encontrar el mapeo apropiado para el resto de esta lectura (flecha verde). La (c) muestra una combinación de estas dos estrategias: el comienzo de la lectura se mapea usando el índice FM global (flecha azul), se extiende hasta que alcanza el final del exón (flecha violeta), se mapea usando el índice FM local (flecha verde) y se extiende de nuevo (flecha violeta).
STAR aligner (Dobin et al. 2013) es una alternativa rápida para mapear lecturas RNA-Seq contra un genoma de referencia utilizando un suffix array sin comprimir. Funciona en dos etapas. En la primera etapa realiza una búsqueda de semillas:
Aquí una lectura se divide entre dos exones consecutivos. STAR empieza a buscar un prefijo mapeable máximo (MMP) desde el principio de la lectura hasta que ya no puede coincidir continuamente. Después de este punto, empieza a buscar un MMP para la parte no coincidente de la lectura (a). En el caso de emparejamientos erróneos (b) y regiones no alineables (c), los MMP sirven como anclas a partir de las cuales extender los alineamientos.
En la segunda etapa STAR stitches MMPs to generate read-level alignments that (contrary to MMPs) can contain mismatches and indels. Se utiliza un esquema de puntuación para evaluar y priorizar las combinaciones de cosido y para evaluar las lecturas que mapean múltiples localizaciones. STAR es extremadamente rápido, pero requiere una cantidad considerable de RAM para funcionar con eficacia.
Mapeo
Mapearemos nuestras lecturas en el genoma de Drosophila melanogaster usando STAR (Dobin et al. 2013).
Práctica: Mapeo empalmado
Importe la anotación de genes Ensembl para Drosophila melanogaster (Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz) de la biblioteca de Datos Compartidos si está disponible o de Zenodo a su actual historial Galaxy
Cambie el nombre del conjunto de datos si es necesario
Compruebe que el tipo de datos es gtf y no gff, y que la base de datos es dm6
Comentario: ¿Cómo obtener el archivo de anotación?
Los archivos de anotación de los organismos modelo pueden estar disponibles en la biblioteca de Datos Compartidos (la ruta a ellos cambiará de un servidor Galaxy a otro). También puede recuperar el archivo de anotación de UCSC (utilizando la herramienta UCSC Main).
Para generar este archivo específico, el archivo de anotación se descargó de Ensembl, que proporciona una base de datos de transcritos más completa, y se adaptó para que funcionara con el genoma dm6, instalado en servidores Galaxy compatibles.
RNA STAR ( Galaxy version 2.7.11a+galaxy0) con los siguientes parámetros para mapear sus lecturas en el genoma de referencia:
“Single-end or paired-end reads “: Paired-end (as collection)
param-collection“RNA-Seq FASTQ/FASTA paired reads “: the Cutadapt on collection N: Reads (output of Cutadapttool)
“Custom or built-in reference genome” “: Use a built-in index
“Reference genome with or without an annotation “: use genome reference without builtin gene-model but provide a gtf
“Select genome reference”: Fly (Drosophila melanogaster): dm6 Full
param-file“Gene model (gff3,gtf) file for splice junctions “: el Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz importado
“Length of the genomic sequence around annotated junctions “: 36
Este parámetro debe ser la longitud de las lecturas - 1
“Per gene/transcript output “: Per gene read counts (GeneCounts)
“Compute coverage “:
Yes in bedgraph format
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los logs STAR con los siguientes parámetros:
En “Results “:
“Results “
“Which tool was used generate logs?”: STAR
En “STAR output “:
param-repeat“Insertar STAR output”
“Type of STAR output? “: Log
param-collection“STAR log output “: RNA STAR on collection N: log (salida de RNA STARtool)
Preguntas
¿Qué porcentaje de lecturas se mapean exactamente una vez para ambas muestras?
¿Cuáles son las otras estadísticas disponibles?
Más del 83% para GSM461177_untreat_paired y del 79% para GSM461180_treat_paired. Podemos continuar con el análisis, ya que sólo los porcentajes inferiores al 70% deben investigarse para detectar una posible contaminación.
También tenemos acceso al número y porcentaje de lecturas que están mapeadas en varias localizaciones, mapeadas en demasiadas localizaciones diferentes, no mapeadas porque son demasiado cortas.
Podríamos haber sido probablemente más estrictos en la longitud mínima de lectura para evitar estas lecturas no mapeadas debido a su longitud.
Según el informe MultiQC, alrededor del 80% de las lecturas de ambas muestras se mapean exactamente una vez con el genoma de referencia. Podemos proseguir con el análisis, ya que sólo los porcentajes inferiores al 70% deben investigarse para detectar una posible contaminación. Ambas muestras tienen un porcentaje bajo (menos del 10%) de lecturas que se corresponden con múltiples ubicaciones en el genoma de referencia. Esto está dentro del rango normal para la secuenciación de lectura corta de Illumina, pero puede ser menor para los nuevos conjuntos de datos de secuenciación de lectura larga que pueden abarcar regiones repetidas más grandes en el genoma de referencia y será mayor para las bibliotecas de extremo 3’.
La salida principal de STAR es un archivo BAM.
Un fichero BAM (Binary Alignment Map) es un archivo binario comprimido que almacena las secuencias leídas, indicando si han sido alineadas a una secuencia de referencia (por ejemplo, un cromosoma) y, en caso afirmativo, la posición en la secuencia de referencia donde han sido alineadas.
Práctica: Inspeccionar un BAM/SAM file
Inspecciona el param-file output de RNA STARtool
Un fichero BAM (o SAM, la versión sin comprimir) consiste de:
Una sección de cabecera (las líneas que comienzan con @) que contiene metadatos, en particular los nombres y longitudes de los cromosomas (líneas que comienzan con el símbolo @SQ).
Una sección de alineamiento que consiste en una tabla con 11 campos obligatorios, además de un número variable de campos opcionales:
Col
Campo
Tipo
Breve descripción
1
QNAME
String
Query template NAME
2
FLAG
Integer
Bitwise FLAG
3
RNAME
String
References sequence NAME
4
POS
Integer
1- based leftmost mapping POSition
5
MAPQ
Integer
MAPping Quality
6
CIGAR
String
CIGAR String
7
RNEXT
String
Ref. name of the mate/next read
8
PNEXT
Integer
Position of the mate/next read
9
TLEN
Integer
Observed Template LENgth
10
SEQ
String
Segment SEQuence
11
QUAL
String
ASCII of Phred-scaled base QUALity+33
Preguntas
¿Qué información se encuentra en un fichero BAM/SAM?
¿Cuál es la información adicional en comparación a un archivo FASTQ?
Información de calidad y de secuencias, como un FASTQ
Información de mapeo, localización de la lectura en el cromosoma, calidad del mapeo, etc.
Inspección de los resultados del mapeo
El archivo BAM contiene información de todas nuestras lecturas, lo que dificulta su inspección y exploración en formato de texto. Una potente herramienta para visualizar el contenido de los archivos BAM es Integrative Genomics Viewer (IGV, Robinson et al. 2011).
Haga clic en la colección RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
Expandir el param-file archivo GSM461177_untreat_paired.
Haga clic en el icono galaxy-barchart visualizar en el bloque de archivo GSM461177.
En el panel central haga clic en local en display with IGV (local, D. melanogaster (dm6)) para cargar las lecturas en el navegador IGV
Comentario
Para que este paso funcione, necesitará tener IGV o Java Web Start instalado en su máquina. Sin embargo, las preguntas de esta sección también pueden responderse inspeccionando las capturas de pantalla de IGV que aparecen a continuación.
Figura 17: Captura de pantalla de un gráfico Sashimi del cromosoma 4
¿Qué representa el gráfico de barras rojas verticales? ¿Y los arcos con números?
¿Qué significan los números de los arcos?
¿Por qué observamos diferentes grupos apilados de cajas azules enlazadas en la parte inferior?
La cobertura de cada pista de alineamiento se representa en un gráfico de barras rojas. Los arcos representan las uniones de empalme observadas, es decir, las lecturas que abarcan intrones.
Los números se refieren al número de lecturas de unión observadas.
Los diferentes grupos de cuadros enlazados en la parte inferior representan los diferentes transcritos de los genes en esta ubicación que están presentes en el archivo GTF.
La calidad de los datos y del mapeo puede comprobarse aún más, por ejemplo, inspeccionando el nivel de duplicación de lecturas, el número de lecturas mapeadas a cada cromosoma, la cobertura del cuerpo génico y la distribución de lecturas a través de las características.
Las lecturas duplicadas pueden provenir de genes altamente expresados, por lo que normalmente se mantienen en el análisis de expresión diferencial RNA-Seq. Pero un alto porcentaje de duplicados puede indicar un problema, por ejemplo, una sobreamplificación durante la PCR de una biblioteca de baja complejidad.
MarkDuplicates de Picard suite examina registros alineados de un archivo BAM para localizar lecturas duplicadas, es decir, lecturas que se mapean en la misma ubicación (basándose en la posición de inicio del mapeo).
Práctica: Comprobar lecturas duplicadas
MarkDuplicates ( Galaxy version 2.18.2.4) con los siguientes parámetros:
param-collection“Seleccionar conjunto de datos SAM/BAM o colección de conjuntos de datos “: RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los logs de MarkDuplicates con los siguientes parámetros:
En “Resultados “:
“Resultados “
“Which tool was used generate logs?”: Picard
En “salida Picard “:
param-repeat“Insertar salida Picard “
“¿Tipo de salida Picard? “: Markdups
param-collection“Picard output “: MarkDuplicates on collection N: MarkDuplicate metrics (salida de MarkDuplicatestool)
Preguntas
¿Cuáles son los porcentajes de lecturas duplicadas para cada muestra?
La muestra GSM461177_untreat_paired tiene un 25,9% de lecturas duplicadas mientras que GSM461180_treat_paired tiene un 27,8%.
En general, obtener hasta un 50% de lecturas duplicadas se considera normal. Por lo tanto, nuestras dos muestras están bien.
Número de lecturas asignadas a cada cromosoma
Para evaluar la calidad de la muestra (por ejemplo, exceso de contaminación mitocondrial), podemos comprobar el sexo de las muestras, o para ver si algún cromosoma tiene genes altamente expresados, podemos comprobar el número de lecturas mapeadas a cada cromosoma utilizando IdxStats de la suite Samtools.
Práctica: Comprobar el número de lecturas asignadas a cada cromosoma
Samtools idxstats ( Galaxy version 2.0.4) con los siguientes parámetros:
param-collection“Archivo BAM “: RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los logs de idxstats con los siguientes parámetros:
En “Resultados “:
“Resultados “
“Which tool was used generate logs?”: Samtools
En “Samtools output “:
param-repeat“Insertar salida Samtools “
“¿Tipo de salida Samtools? “: idxstats
param-collection“Samtools idxstats output “: Samtools idxstats on collection N (salida de Samtools idxstatstool)
Preguntas
¿Cuántos cromosomas tiene el genoma de Drosophila?
¿Dónde se encuentran la mayoría de las lecturas?
¿Podemos determinar el sexo de las muestras?
El genoma de Drosophila tiene 4 pares de cromosomas: X/Y, 2, 3 y 4.
Las lecturas corresponden principalmente al cromosoma 2 (chr2L y chr2R), 3 (chr3L y chr3R) y X. Sólo unas pocas lecturas corresponden al cromosoma 4, lo que es de esperar dado que este cromosoma es muy pequeño.
A juzgar por el porcentaje de lecturas X+Y, la mayoría de las lecturas se asignan a X y sólo unas pocas a Y. Esto indica que probablemente no hay muchos genes en Y, por lo que las muestras son probablemente femeninas.
Cobertura del cuerpo genético
Las diferentes regiones de un gen forman el cuerpo del gen. Es importante comprobar si la cobertura de lectura es uniforme en todo el cuerpo del gen. Por ejemplo, un sesgo hacia el extremo 5’ de los genes podría indicar la degradación del ARN. Alternativamente, un sesgo hacia el extremo 3’ podría indicar que los datos proceden de un ensayo 3’. Para evaluar esto, podemos utilizar la herramienta Gene Body Coverage del conjunto de herramientas RSeQC (Wang et al. 2012). Esta herramienta escala todas las transcripciones a 100 nucleótidos (utilizando un archivo de anotación proporcionado) y calcula el número de lecturas que cubren cada posición de nucleótido (escalada). Como esta herramienta es muy lenta, calcularemos la cobertura sólo con 200.000 lecturas aleatorias.
Práctica: Comprobar la cobertura del cuerpo del gen
Samtools view ( Galaxy version 1.15.1+galaxy0) con los siguientes parámetros:
param-collection“Conjunto de datos SAM/BAM/CRAM “: mapped_reads (salida de RNA STARtool)
“¿Qué te gustaría mirar?: A filtered/subsampled selection of reads
En “Configurar submuestreo “:
“Subsample alignment “: Specify a target # of reads
“Objetivo # de lecturas “: 200000
“Semilla para el generador de números aleatorios “: 1
“¿Qué le gustaría que se informara? “: All reads retained after filtering and subsampling
“Output format “: BAM (-b)
“Usar una secuencia de referencia “: No
Convert GTF to BED12 ( Galaxy version 357) para convertir el archivo GTF a BED:
param-file“GTF File to convert “: Drosophila_melanogaster.BDGP6.32.109.gtf.gz
Gene Body Coverage (BAM) ( Galaxy version 5.0.1+galaxy2) con los siguientes parámetros:
“Ejecutar cada muestra por separado, o combinar varias muestras en un gráfico “: Run each sample separately
param-collection“Input .bam file “: salida de la vista Samtoolstool
param-file *“Reference gene model “: Convert GTF to BED12 on data N: BED12 (salida de Convert GTF to BED12tool)
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los resultados RSeQC con los siguientes parámetros:
En “Resultados “:
“Resultados “
“Which tool was used generate logs?”: RSeQC
En “RSeQC output “:
param-repeat“Insertar salida RSeQC “
“¿Tipo de salida RSeQC? “: gene_body_coverage
param-collection“RSeQC gene_body_coverage output “: Gene Body Coverage (BAM) on collection N (text) (salida de Gene Body Coverage (BAM)tool)
Preguntas
¿Cómo es la cobertura a través de los cuerpos génicos? ¿Las muestras están sesgadas en 3’ o 5’?
En ambas muestras hay una cobertura bastante uniforme de los extremos 5’ a 3’ (a pesar de algo de ruido en el medio). Así que no hay sesgo obvio en ambas muestras.
Distribución de las lecturas por características
Con los datos de RNA-Seq, esperamos que la mayoría de las lecturas correspondan a exones en lugar de a intrones o regiones intergénicas. Antes de seguir adelante con el recuento y el análisis de expresión diferencial, puede ser interesante comprobar la distribución de las lecturas en las características conocidas de los genes (exones, CDS, 5’ UTR, 3’ UTR, intrones, regiones intergénicas). Por ejemplo, un número elevado de lecturas en regiones intergénicas puede indicar la presencia de contaminación por ADN.
Aquí utilizaremos la herramienta Read Distribution del conjunto de herramientas RSeQC (Wang et al. 2012), que utiliza el archivo de anotación para identificar la posición de las diferentes características de los genes.
Práctica: Comprobar el número de lecturas asignadas a cada cromosoma
Read Distribution ( Galaxy version 5.0.1+galaxy2) con los siguientes parámetros:
param-collection“Archivo .bam/.sam de entrada “: RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
param-file *“Reference gene model “: Archivo BED12 (salida de Convert GTF to BED12tool)
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los resultados de la Distribución de Lecturas con los siguientes parámetros:
En “Resultados “:
“Resultados “
“Which tool was used generate logs?”: RSeQC
En “RSeQC output “:
param-repeat“Insertar salida RSeQC “
“¿Tipo de salida RSeQC? “: read_distribution
param-collection“RSeQC read_distribution output “: Read Distribution on collection N (salida de Distribución de lecturastool)
Preguntas
¿Qué opina de la distribución de las lecturas?
La mayoría de las lecturas están mapeadas a exones (>80%), sólo ~2% a intrones y ~5% a regiones intergénicas, que es lo que esperamos. Esto confirma que nuestros datos son datos RNA-Seq y que el mapeo fue exitoso.
Ahora que hemos comprobado los resultados del mapeo de lecturas, podemos pasar a la siguiente fase del análisis.
Después del mapeo, ahora tenemos la información de dónde están localizadas las lecturas en el genoma de referencia y cómo de bien fueron mapeadas. El siguiente paso en el análisis de datos RNA-Seq es la cuantificación del número de lecturas mapeadas a características genómicas (genes, transcritos, exones, …).
Comentario
La cuantificación depende tanto del genoma de referencia (el archivo FASTA) como de sus anotaciones asociadas (el archivo GTF). Es extremadamente importante utilizar un archivo de anotaciones que corresponda a la misma versión del genoma de referencia que utilizó para el mapeo (por ejemplo, dm6 aquí), ya que las coordenadas cromosómicas de los genes suelen ser diferentes entre las distintas versiones del genoma de referencia.
Aquí nos centraremos en los genes, ya que nos gustaría identificar los que se expresan diferencialmente debido al knockdown del gen Pasilla.
Recuento del número de lecturas por gen anotado
Para comparar la expresión de genes individuales entre diferentes condiciones (e.g. con o sin depleción de PS), un primer paso esencial es cuantificar el número de lecturas por gen, o más específicamente el número de lecturas mapeadas a los exones de cada gen.
Figura 19: Recuento del número de lecturas por gen anotado
Preguntas
En la imagen anterior,
¿Cuántas lecturas se encuentran para los diferentes exones?
¿Cuántas lecturas se encuentran para los diferentes genes?
Número de lecturas por exón
Exon
Number of reads
gene1 - exon1
3
gene1 - exon2
2
gene2 - exon1
3
gene2 - exon2
4
gene2 - exon3
3
el gen1 tiene 4 lecturas, no 5, debido al empalme de la última lectura (gen1 - exón1 + gen1 - exón2). el gen2 tiene 6 lecturas, 3 de las cuales están empalmadas.
Existen dos herramientas principales para el recuento de lecturas: HTSeq-count (Anders et al. 2015) o featureCounts (Liao et al. 2013). Además, STAR permite contar lecturas mientras se mapea: sus resultados son idénticos a los de HTSeq-count. Mientras que esta salida es suficiente para la mayoría de los análisis, featureCounts ofrece más personalización sobre cómo contar lecturas (calidad mínima de mapeo, contar lecturas en lugar de fragmentos, contar transcripciones en lugar de genes, etc.).
En principio, el recuento de lecturas que se solapan con características genómicas es una tarea bastante sencilla. Pero es necesario determinar la estridencia de la biblioteca. De hecho, este es un parámetro de featureCounts. Por el contrario, STAR evalúa los recuentos en los tres posibles strandnesses pero usted todavía necesita esta información para extraer los recuentos que corresponden a su biblioteca.
Estimación de la strandness
Los ARN a los que normalmente se dirigen los experimentos de ARN-Seq son monocatenarios (por ejemplo, ARNm) y, por lo tanto, tienen polaridad (extremos 5’ y 3’ funcionalmente distintos). Durante un experimento típico de RNA-Seq, la información sobre la cadena se pierde después de que ambas cadenas de cDNA se sinteticen, se seleccionen por tamaño y se conviertan en una biblioteca de secuenciación. Sin embargo, esta información puede ser muy útil para el paso de recuento de lecturas, especialmente para lecturas localizadas en el solapamiento de 2 genes que están en hebras diferentes.
Figura 20: Si la información de estrangulamiento se perdió durante la preparación de la biblioteca, la lectura1 se asignará al gen1 localizado en la cadena directa, pero la lectura2 será 'ambigua', ya que puede asignarse al gen1 (cadena directa) o al gen2 (cadena inversa).
Algunos protocolos de preparación de bibliotecas crean las denominadas bibliotecas de ARN-Seq stranded que conservan la información de la cadena (Levin et al. 2010 ofrece una excelente descripción general). En la práctica, con los protocolos de RNA-Seq de Illumina es poco probable que se encuentre con todas las posibilidades descritas en este artículo. Lo más probable es que se encuentre con
Datos de ARN-Seq sin cadena
Datos de RNA-Seq trenzados generados por el uso de kits especializados de aislamiento de RNA durante la preparación de la muestra
Figura 21: Relación entre la orientación del ADN y el ARN
La implicación de RNA-Seq stranded es que se puede distinguir si las lecturas derivan de transcritos codificados hacia delante o hacia atrás. En el siguiente ejemplo, los recuentos para el gen Mrpl43 sólo pueden estimarse eficientemente en una librería stranded ya que la mayor parte se solapa con el gen Peo1 en la orientación inversa:
Figura 22: Non-stranded (top) vs. reverse strand-specific (bottom) RNA-Seq read alignment (using IGV, forward mapping reads are red and reverse mapping reads are blue )
Dependiendo del enfoque, y de si se realiza secuenciación de extremo único o de extremo pareado, existen múltiples posibilidades sobre cómo interpretar los resultados del mapeo de estas lecturas con el genoma:
Figura 23: Efectos de los tipos de bibliotecas RNA-Seq (Figura adaptada de la documentación de Sailfish)
Esta información debería estar incluida en los archivos FASTQ, ¡pregunte en su centro de secuenciación! Si no es así, intente encontrarla en el sitio donde descargó los datos o en la publicación correspondiente.
Figura 24: En una biblioteca de hebra directa, las lecturas se mapean principalmente en la misma hebra que los genes. En una biblioteca inversa trenzada, las lecturas se sitúan principalmente en la cadena opuesta. Con una biblioteca no trenzada, las lecturas se mapean en los genes en ambas cadenas independientemente de la orientación del gen (Ejemplo para una biblioteca de lectura de un solo extremo).
Hay 4 formas de estimar la estridencia a partir de los resultados de STAR (elija la que prefiera)
Podemos hacer una inspección visual de las hebras de lectura en IGV (para conjuntos de datos Paired-end es menos fácil que con una sola lectura y cuando se tienen muchas muestras, esto puede ser doloroso).
Práctica: Estimar el estrangulamiento con IGV para una biblioteca de extremos pareados
Vuelva a su sesión IGV con el GSM461177_untreat_paired BAM abierto.
No hay problema, sólo hay que rehacer los pasos anteriores:
Iniciar IGV localmente
Haga clic en la colección RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
Expandir el param-file archivo GSM461177_untreat_paired.
Haga clic en local en display with IGV local D. melanogaster (dm6) para cargar las lecturas en el navegador IGV
IGVtool
Zoom a chr3R:9,445,000-9,448,000 (Cromosoma 3 entre 9,445 kb a 9,448 kb), en la pista mapped.bam
Haga clic con el botón derecho y seleccione Color Aligments by -> first-in-pair strand
Haga clic con el botón derecho y seleccione Squished
Figura 26: Captura de pantalla del IGV para no hebra (arriba) frente a hebra específica inversa (abajo)
Observe que no hay ninguna lectura en el grupo POSITIVO para la cadena inversa específica.
Alternativamente, en lugar de utilizar el BAM se puede utilizar la cobertura de hebra generada por STAR. Usando pyGenomeTracks podremos visualizar la cobertura en cada hebra para cada muestra. Esta herramienta tiene un montón de parámetros para personalizar sus gráficos.
Práctica: Estimar el strandness con pyGenometracks a partir de la cobertura STAR
pyGenomeTracks ( Galaxy version 3.8+galaxy2):
“Region of the genome to plot “: chr4:540,000-560,000
En "”Include tracks in your plot “:
param-repeat“Insert Include tracks in your plot “
“Choose style of the track “: Bedgraph track
“Plot title “: Deje este campo vacío para que el título del gráfico sea el nombre de la muestra.
param-collection“Track file(s) bedgraph format “: Seleccione RNA STAR on collection N: Coverage Uniquely mapped strand 1.
“Color of track “: Seleccione un color de su elección, por ejemplo azul
“Minimum value “: 0
“height “: 3
“Show visualization of data range”: Yes
param-repeat“Insert Include tracks in your plot “
“Choose style of the track “: Bedgraph track
“Plot title “: Deje este campo vacío para que el título del gráfico sea el nombre de la muestra.
param-collection“Track file(s) bedgraph format “: Seleccione RNA STAR on collection N: Coverage Uniquely mapped strand 2.
“Color of track “: Seleccione un color de su elección distinto del primero, por ejemplo rojo
“Minimum value “: 0
“height “: 3
“Show visualization of data range “: Yes
param-repeat“Insert Include tracks in your plot “
“Choose style of the track “: Gene track / Bed track
“Plot title “: Genes
param-file“Track file(s) bed or gtf format “: Seleccione Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
Figura 28: Cobertura STAR para el strand 1 en azul y el strand 2 en rojo para librerías unstranded y reverse stranded
Observe que la cobertura en la hebra 1 es muy baja para la muestra stranded_PE mientras que el gen es forward. Esto significa que la biblioteca de stranded_PE es de cadena inversa. Por el contrario, para unstranded_PE la escala es comparable para ambas cadenas.
Se puede utilizar la salida de STAR con los recuentos. En efecto, como se ha explicado antes, STAR evalúa el número de lecturas en los genes para los tres escenarios posibles: librería no trenzada, trenzado directo o trenzado inverso. La condición que atribuye más lecturas al gen debe ser la condición que corresponde a su biblioteca.
Práctica: Estimar el estrangulamiento con recuentos STAR
MultiQC ( Galaxy version 1.11+galaxy1) para agregar los recuentos STAR con los siguientes parámetros:
En “Results “:
“Results “
“Which tool was used generate logs?”: STAR
En “STAR output “:
param-repeat“Insert STAR output “
“Type of STAR output? “: Gene counts
param-collection“STAR gene count output “: RNA STAR on collection N: reads per gene (salida de RNA STARtool)
Preguntas
¿Qué porcentaje de lecturas se asignan a genes si la biblioteca es no trenzada/misma trenzada/trenzada inversa?
Figura 32: Recuento de genes no trenzados para bibliotecas no trenzadas y trenzadas inversasOpen image in new tab
Figura 33: Gene counts same stranded for unstranded and reverse stranded libraryOpen image in new tab
Figura 34: Gene counts reverse stranded for unstranded and reverse stranded library
Observe que hay muy pocas lecturas atribuidas a genes para la misma cadena. Las cifras son comparables entre las bibliotecas no trenzadas y las trenzadas inversas, ya que muy pocos genes se solapan en las cadenas opuestas, pero aún así la cifra pasa del 63,6% (no trenzadas) al 65% (trenzadas inversas).
Otra opción es estimar estos parámetros con una herramienta llamada Infer Experiment del conjunto de herramientas RSeQC (Wang et al. 2012).
Esta herramienta toma los archivos BAM del mapeo, selecciona una submuestra de las lecturas y compara sus coordenadas genómicas y hebras con las del Reference gene model “: (de un archivo de anotación). Basándose en la hebra de los genes, puede determinar si la secuenciación es específica de la hebra y, en caso afirmativo, cómo son las hebras de las lecturas (directa o inversa).
Práctica: Determinación de la cadena de la biblioteca mediante el Experimento Infer
Convert GTF to BED12 ( Galaxy version 357) para convertir el archivo GTF a BED:
param-file“GTF File to convert “: Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
Es posible que ya haya convertido este archivo BED12 a partir del conjunto de datos Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz anteriormente si realizó la parte detallada sobre las comprobaciones de calidad. En este caso, no es necesario volver a hacerlo una segunda vez
Infer Experiment ( Galaxy version 5.0.3+galaxy0) para determinar la robustez de la biblioteca con los siguientes parámetros:
param-collection“Input .bam file “: RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
param-file“Reference gene model “: Archivo BED12 (salida de Convert GTF to BED12tool)
“Number of reads sampled “: 200000
Infer Experiment ( Galaxy version 5.0.3+galaxy0) tool genera un archivo con información sobre:
Biblioteca de extremo pareado o de extremo único
Fracción de lecturas que no se han podido determinar
2 líneas
Para extremo único
Fraction of reads explained by "++,--": fracción de lecturas asignadas a la cadena directa
Fraction of reads explained by "+-,-+": fracción de lecturas asignadas a la cadena inversa
Para los extremos pareados
Fraction of reads explained by "1++,1--,2+-,2-+": fracción de lecturas asignadas a la cadena directa
Fraction of reads explained by "1+-,1-+,2++,2--": fracción de lecturas asignadas a la cadena inversa
Si los dos números de “Fracción de lecturas explicadas por” están próximos entre sí, concluimos que la biblioteca no es un conjunto de datos de cadena específica (o no cadena).
Preguntas
¿Cuáles son los resultados de “Fracción de las lecturas explicada por” para GSM461177_untreat_paired?
¿Cree que el tipo de biblioteca de las 2 muestras es trenzada o no trenzada?
Resultados para GSM461177_untreat_paired:
Comentario: Los resultados pueden variar
Tus resultados pueden ser ligeramente diferentes de los presentados en este tutorial debido a las diferentes versiones de las herramientas, datos de referencia, bases de datos externas, o debido a procesos estocásticos en los algoritmos.
This is PairEnd Data
Fraction of reads failed to determine: 0.1013
Fraction of reads explained by "1++,1--,2+-,2-+": 0.4626
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4360
de modo que el 46,26% de las lecturas se asignan a la cadena directa y el 43,60% a la cadena inversa.
Se encuentran estadísticas similares para GSM461180_treat_paired, por lo que la biblioteca parece ser del tipo unstranded para ambas muestras.
Comentario: ¿Cómo sería si la biblioteca estuviera trenzada?
Siguiendo con el ejemplo de 2 BAM, obtenemos para el no trenzado
This is PairEnd Data
Fraction of reads failed to determine: 0.0382
Fraction of reads explained by "1++,1--,2+-,2-+": 0.4847
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4771
Y para la cadena inversa:
This is PairEnd Data
Fraction of reads failed to determine: 0.0504
Fraction of reads explained by "1++,1--,2+-,2-+": 0.0061
Fraction of reads explained by "1+-,1-+,2++,2--": 0.9435
Como a veces es bastante difícil averiguar qué ajustes corresponden a los de otros programas, la siguiente tabla puede ser útil para identificar el tipo de biblioteca:
Library type
Infer Experiment
TopHat
HISAT2
HTSeq-count
featureCounts
Paired-End (PE) - SF
1++,1–,2+-,2-+
FR Second Strand
Second Strand F/FR
yes
Forward (1)
PE - SR
1+-,1-+,2++,2–
FR First Strand
First Strand R/RF
reverse
Reverse (2)
Single-End (SE) - SF
++,–
FR Second Strand
Second Strand F/FR
yes
Forward (1)
SE - SR
+-,-+
FR First Strand
First Strand R/RF
reverse
Reverse (2)
PE, SE - U
undecided
FR Unstranded
default
no
Unstranded (0)
Recuento de lecturas por genes
Hands-on: Choose Your Own Tutorial
Esto es una sección 'Choose Your Own Tutorial' (CYOT) (también conocida como 'Choose Your Own Analysis' (CYOA)), donde puedes elegir entre varios caminos. Haz click en alguno de los botones debajo para elegir qué quieres seguir en este tutorial
Para contar el número de lecturas por gen, ofrecemos un tutorial paralelo para los 2 métodos (STAR y featureCounts) que dan resultados muy similares. ¿Qué método prefiere utilizar?
Como usted eligió utilizar la opción featureCounts del tutorial, ahora ejecutamos featureCounts para contar el número de lecturas por gen anotado.
Práctica: Contando el número de lecturas por gen anotado
featureCounts ( Galaxy version 2.0.3+galaxy2) con los siguientes parámetros para contar el número de lecturas por gen:
param-collection“Alignment file “: RNA STAR on collection N: mapped.bam (salida de RNA STARtool)
“Specify strand information “: Unstranded
“Gene annotation file “: A GFF/GTF file in your history
Algunas lecturas no han sido asignadas porque estaban mapeadas; otras no han sido asignadas a ninguna característica o han sido asignadas a características ambiguas.
Si el porcentaje es inferior al 50%, debería investigar dónde se están mapeando sus lecturas (dentro de genes o no, con IGV) y comprobar que la anotación corresponde a la versión correcta del genoma de referencia.
La salida principal de featureCounts es una tabla con los recuentos, es decir, el número de lecturas (o fragmentos en el caso de lecturas paired-end) mapeadas a cada gen (en filas, con su ID en la primera columna) en la anotación proporcionada. FeatureCount genera también los conjuntos de datos de salida feature length. Necesitaremos este archivo más adelante cuando ejecutemos la herramienta goseq.
Como usted eligió usar la versión STAR del tutorial, usaremos STAR para contar las lecturas.
Como se ha escrito anteriormente, durante el mapeo, STAR contó las lecturas para cada gen proporcionado en el fichero de anotación de genes (esto se consiguió mediante la opción Per gene read counts (GeneCounts)). Sin embargo, esta salida proporciona algunas estadísticas al principio y los recuentos para cada gen dependiendo de la biblioteca (unstranded es la columna 2, stranded forward es la columna 3 y stranded reverse es la columna 4).
Práctica: Inspeccionar la salida de STAR
Inspeccionar los recuentos de GSM461177_untreat_paired en la colección RNA STAR on collection N: reads per gene
Preguntas
¿Cuántas lecturas están sin mapear/multi-mapeadas?
¿En qué línea comienza el recuento de genes?
¿Cuáles son las diferentes columnas?
¿Qué columnas son las más interesantes para nuestro conjunto de datos?
Hay 1.190.029 lecturas no mapeadas y 571.324 lecturas multimapeadas.
Comienza en la línea 5 con el gen FBgn0250732.
Hay 4 columnas:
ID del gen
Recuentos para RNA-seq no trenzado
Recuentos de la primera cadena de lectura alineada con el ARN
Recuentos de la 2ª cadena de lectura alineada con el ARN
Necesitamos la columna Gene ID y la 2ª columna debido a la falta de coherencia de nuestros datos
Reformatearemos la salida de STAR para que sea similar a la salida de featureCounts (u otros programas de recuento) que sólo tiene 2 columnas, una con IDs y la otra con recuentos.
Práctica: Reformateo de la salida STAR
Select last ( Galaxy version 1.1.0) líneas de un conjunto de datos (tail) para eliminar las 4 primeras líneas con los siguientes parámetros:
param-collection“Text file”: RNA STAR on collection N: reads per gene (salida de RNA STARtool)
“Operation “: Keep everything from this line on
“Number of lines “: 5
Cut columnas de una tabla con los siguientes parámetros:
“Cut columns “: c1,c2
“Delimited by “: Tab
param-collection“De “: Select last on collection N (salida de Select lasttool)
Cambiar el nombre de la colección FeatureCount-like files
Más adelante en el tutorial necesitaremos obtener el tamaño de cada gen. Esta es una de las salidas de FeatureCounts pero también podemos obtenerla directamente del fichero de anotación del gen. Como esto es bastante largo, recomendamos lanzarlo ahora.
Práctica: Cómo obtener la longitud del gen
Gene length and GC content ( Galaxy version 0.1.2) con los siguientes parámetros:
“Select a built-in GTF file or one from your history “: Use a GTF from history
param-file“Select a GTF file “: Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
“Analysis to perform “: gene lengths only
¡Precaucion!: Compruebe la versión de la herramienta a continuación
Esto sólo funcionará con la versión 0.1.2 o superior
Las herramientas se actualizan con frecuencia a nuevas versiones. Su Galaxy puede tener varias versiones disponibles de la misma herramienta. Por defecto, se le mostrará la última versión de la herramienta.
Pasar a otra versión de una herramienta:
Abrir la herramienta
Haga clic en el logo tool-versions versiones arriba a la derecha**
Seleccione la versión deseada en la lista desplegable
Preguntas
¿Qué característica tiene más recuentos en ambas muestras? (Sugerencia: utilice la herramienta Ordenar)
Para mostrar la característica detectada más abundante, debemos ordenar la tabla de recuentos. Esto se puede hacer de la siguiente manera:
Sort ( Galaxy version 1.1.1) con los siguientes parámetros:
param-collection“Sort Query “: featureCounts on collection N: Counts (salida de featureCountstool)Utilizar la colección FeatureCount-like files
“Number of header “: 10
En “1: Selecciones de columna “:
“on column “: Column: 2
Esta columna contiene el número de lecturas = counts
“in “: Descending order
Inspeccionar el resultado
El resultado de ordenar la tabla en la columna 2 revela que FBgn0284245 es la característica con más recuentos (alrededor de 128.740 en GSM461177_untreat_paired y 127.400 en GSM461180_treat_paired).
Comparar diferentes archivos de salida es más fácil si podemos ver más de un conjunto de datos simultáneamente. La función Scratchbook nos permite crear una colección de conjuntos de datos que se mostrarán juntos en la pantalla.
Práctica: (Opcional) Ver los recuentos ordenados utilizando el Scratchbook
El Scratchbook se activa haciendo clic en el icono de nueve bloques que aparece a la derecha de la barra de menú superior de Galaxy:
Figura 37: Barra de menú con el icono de Scratchbook activado
Haga clic en el galaxy-eye (ojo) para ver uno de los archivos de conteos clasificados. En lugar de ocupar toda la barra central, la vista del conjunto de datos se muestra ahora superpuesta:
Figura 38: Scratchbook muestra un conjunto de datos superpuesto
A continuación, haga clic en el galaxy-eye (ojo) en el segundo archivo de recuentos ordenados. El segundo conjunto de datos pasa por encima del primero, pero puede mover la ventana para ver los dos conjuntos de datos uno al lado del otro:
Figura 39: Scratchbook mostrando dos conjuntos de datos uno al lado del otro
Para salir del modo de selección Scratchbook, haga clic de nuevo en el icono Scratchbook. Puede decidir cerrar las ventanas o reducirlas para visualizarlas más tarde.
Aquí contamos las lecturas asignadas a genes de dos muestras. Es muy interesante volver a realizar el mismo procedimiento en los otros conjuntos de datos, especialmente para comprobar cómo difieren los parámetros dado el diferente tipo de datos (single-end frente a paired-end).
Práctica: (Opcional) Reejecutar en los otros conjuntos de datos
Puede realizar el mismo proceso en los otros archivos de secuencia disponibles en Zenodo y en la biblioteca de datos.
Datos por pares
GSM461178_1 y GSM461178_2 que se pueden etiquetar como GSM461178_untreat_paired
GSM461181_1 y GSM461181_2 que se pueden etiquetar como GSM461181_treat_paired
Datos de extremo único
GSM461176 que puede etiquetar GSM461176_untreat_single
GSM461179 que puede etiquetar GSM461179_treat_single
GSM461182 que puede etiquetar GSM461182_untreat_single
Los enlaces a estos archivos se encuentran a continuación:
Para los datos de un solo extremo, no es necesario aplanar la colección antes del paso Falco. Los parámetros de todas las herramientas son los mismos excepto STAR para el que puede establecer Length of the genomic sequence around annotated junctions a 74 ya que un conjunto de datos tiene lecturas de 75bp (otros son 44bp y 45bp) y FeatureCount si sus datos ya no están emparejados.
Análisis de la expresión génica diferencial
Identificación de los rasgos expresados diferencialmente
Para poder identificar la expresión génica diferencial inducida por la depleción de PS, todos los conjuntos de datos (3 tratados y 4 no tratados) deben analizarse siguiendo el mismo procedimiento. Para ahorrar tiempo, hemos ejecutado los pasos anteriores por usted. Obtenemos entonces 7 ficheros con los recuentos de cada gen de Drosophila para cada muestra.
Práctica: Importar todos los ficheros de recuento
Crear un nuevo historial vacío
Haz click sobre el icono new-history en la parte superior del panel de historiales.
Importar los siete archivos de recuento de Zenodo o la biblioteca de Datos Compartidos:
Se podría pensar que podemos comparar directamente los valores de recuento de los archivos y calcular el grado de expresión génica diferencial. Sin embargo, no es tan sencillo.
Imaginemos que tenemos recuentos de RNA-Seq de 3 muestras para un genoma con 4 genes:
Gene
Sample 1 Counts
Sample 2 Counts
Sample 3 Counts
A (2kb)
10
12
30
B (4kb)
20
25
60
C (1kb)
5
8
15
D (10kb)
0
0
1
La muestra 3 tiene más lecturas que las otras réplicas, independientemente del gen. Tiene una profundidad de secuenciación mayor que las otras réplicas. El gen B es el doble de largo que el gen A: podría explicar por qué tiene el doble de lecturas, independientemente de las réplicas.
Por lo tanto, el número de lecturas secuenciadas asignadas a un gen depende de:
La profundidad de secuenciación de las muestras
Las muestras secuenciadas con mayor profundidad tendrán más lecturas asignadas a cada gen
La longitud del gen
Los genes más largos tendrán más lecturas asignadas a ellos
Para comparar muestras o expresiones génicas, es necesario normalizar los recuentos de genes. Podríamos utilizar TPM (Transcritos por Kilobase Millón).
Estas tres métricas se utilizan para normalizar las tablas de recuento de:
profundidad de secuenciación (la parte del “Millón”)
longitud del gen (la parte “Kilobase”)
Utilicemos el ejemplo anterior para explicar RPKM, FPKM y TPM.
Para RPKM (lecturas por kilobase millón),
Calcule el factor de escala “por millón”: sume el total de lecturas de una muestra y divida ese número por 1.000.000.
Gene
Sample 1 Counts
Sample 2 Counts
Sample 3 Counts
A (2kb)
10
12
30
B (4kb)
20
25
60
C (1kb)
5
8
15
D (10kb)
0
0
1
Total reads
35
45
106
Scaling factor
3.5
4.5
10.6
Debido a los pequeños valores del ejemplo, utilizamos “por decenas” en lugar de “por millones” y, por tanto, dividimos la suma por 10 en lugar de por 1.000.000.
Dividir el número de lecturas por el factor de escala “por millón
Esto normaliza la profundidad de secuenciación, dando lecturas por millón (RPM)
Gene
Sample 1 RPM
Sample 2 RPM
Sample 3 RPM
A (2kb)
2.86
2.67
2.83
B (4kb)
5.71
5.56
5.66
C (1kb)
1.43
1.78
1.43
D (10kb)
0
0
0.09
En el ejemplo utilizamos el factor de escala “por decenas” y obtenemos lecturas por decenas
Divida los valores de RPM por la longitud del gen, en kilobases.
Gene
Sample 1 RPKM
Sample 2 RPKM
Sample 3 RPKM
A (2kb)
1.43
1.33
1.42
B (4kb)
1.43
1.39
1.42
C (1kb)
1.43
1.78
1.42
D (10kb)
0
0
0.009
FPKM (Fragmentos por Kilobase Millón) es muy similar a RPKM. RPKM se utiliza para RNA-seq de extremo único, mientras que FPKM se utiliza para RNA-seq de extremo pareado. En el caso del extremo único, cada lectura corresponde a un único fragmento secuenciado. Con RNA-seq paired-end, dos lecturas de un par se mapean a partir de un único fragmento, o si una lectura del par no se mapeó, una lectura puede corresponder a un único fragmento (en caso de que decidiéramos conservarlas). FPKM mantiene un registro de los fragmentos de forma que un fragmento con 2 lecturas se cuenta sólo una vez.
TPM (Transcripciones por Kilobase Millón) es muy similar a RPKM y FPKM, excepto en el orden de la operación
Divida el número de lecturas por la longitud de cada gen en kilobases
Esto da las lecturas por kilobase (RPK).
Gene
Sample 1 RPK
Sample 2 RPK
Sample 3 RPK
A (2kb)
5
6
15
B (4kb)
5
6.25
15
C (1kb)
5
8
15
D (10kb)
0
0
0.1
Calcular el factor de escala “por millón”: sumar todos los valores RPK de una muestra y dividir este número por 1.000.000
Gene
Sample 1 RPK
Sample 2 RPK
Sample 3 RPK
A (2kb)
5
6
15
B (4kb)
5
6.25
15
C (1kb)
5
8
15
D (10kb)
0
0
0.1
Total RPK
15
20.25
45.1
Scaling factor
1.5
2.03
4.51
Como en el caso anterior, debido a los pequeños valores del ejemplo, utilizamos “por decenas” en lugar de “por millones” y, por lo tanto, dividimos la suma por 10 en lugar de por 1.000.000.
Dividir los valores RPK por el factor de escala “por millón
Gene
Sample 1 TPM
Sample 2 TPM
Sample 3 TPM
A (2kb)
3.33
2.96
3.33
B (4kb)
3.33
3.09
3.33
C (1kb)
3.33
3.95
3.33
D (10kb)
0
0
0.1
A diferencia de RPKM y FPKM, al calcular TPM, normalizamos primero la longitud del gen y luego la profundidad de secuenciación. Sin embargo, los efectos de esta diferencia son bastante profundos, como ya vimos con el ejemplo.
Las sumas de cada columna son muy diferentes:
RPKM
Gene
Sample 1 RPKM
Sample 2 RPKM
Sample 3 RPKM
A (2kb)
1.43
1.33
1.42
B (4kb)
1.43
1.39
1.42
C (1kb)
1.43
1.78
1.42
D (10kb)
0
0
0.009
Total
4.29
4.5
4.25
TPM
Gene
Sample 1 TPM
Sample 2 TPM
Sample 3 TPM
A (2kb)
3.33
2.96
3.33
B (4kb)
3.33
3.09
3.33
C (1kb)
3.33
3.95
3.33
D (10kb)
0
0
0.1
Total
10
10
10
La suma de todos los TPM en cada muestra es la misma. Esto facilita la comparación de la proporción de lecturas que corresponden a un gen en cada muestra. Por el contrario, con RPKM y FPKM, la suma de las lecturas normalizadas en cada muestra puede ser diferente, y esto hace más difícil comparar muestras directamente.
En el ejemplo, el TPM para el gen A en la muestra 1 es de 3,33 y en la muestra 2 es de 3,33. La misma proporción del total de lecturas se asigna entonces al gen A en ambas muestras (0,33 aquí). De hecho, la suma de los TPM en ambas muestras suma el mismo número (10 aquí), el denominador requerido para calcular las proporciones es entonces el mismo independientemente de la muestra, y por lo tanto la proporción de lecturas para el gen A (3.33/10 = 0.33) para ambas muestras.
Con RPKM o FPKM, es más difícil comparar la proporción de lecturas totales porque la suma de lecturas normalizadas en cada muestra puede ser diferente (4,29 para la Muestra 1 y 4,25 para la Muestra 2). Por lo tanto, si el RPKM para el gen A en la Muestra 1 es 1,43 y en la Muestra B es 1,43, no sabemos si la misma proporción de lecturas en la Muestra 1 corresponden al gen A que en la Muestra 2.
Dado que en RNA-Seq se trata de comparar la proporción relativa de lecturas, TPM parece más apropiado que RPKM/FPKM.
RNA-Seq se utiliza a menudo para comparar un tipo de tejido con otro, por ejemplo, músculo frente a tejido epitelial. Y puede ser que haya muchos genes específicos del músculo transcritos en el músculo pero no en el tejido epitelial. A esto lo llamamos diferencia en la composición de la biblioteca.
También es posible observar una diferencia en la composición de la biblioteca en el mismo tipo de tejido tras la eliminación de un factor de transcripción.
Imaginemos que tenemos recuentos de RNA-Seq de 2 muestras (mismo tamaño de biblioteca: 635 reads), para un genoma con 6 genes. Los genes tienen la misma expresión en ambas muestras, excepto uno: sólo la Muestra 1 transcribe el gen D, a un nivel alto (563 reads). Como el tamaño de la biblioteca es el mismo para ambas muestras, la muestra 2 tiene 563 lecturas adicionales que se distribuirán entre los genes A, B, C, E y F.
Gene
Sample 1
Sample 2
A
30
235
B
24
188
C
0
0
D
563
0
E
5
39
F
13
102
Total
635
635
Como resultado, el recuento de lecturas para todos los genes excepto para los genes C y D es realmente alto en la Muestra 2. No obstante, el único gen expresado diferencialmente es el gen D.
TPM, RPKM o FPKM no tienen en cuenta estas diferencias en la composición de la biblioteca durante la normalización, pero herramientas más complejas, como DESeq2, sí lo hacen.
DESeq2 (Love et al. 2014) es una gran herramienta para tratar con datos de ARN-seq y ejecutar análisis de Expresión Génica Diferencial (EGD). Toma archivos de conteo de lecturas de diferentes muestras, los combina en una gran tabla (con los genes en las filas y las muestras en las columnas) y aplica normalización para profundidad de secuenciación y composición de la biblioteca. No necesitamos tener en cuenta la normalización de la longitud de los genes porque estamos comparando los recuentos entre grupos de muestras para el mismo gen.
Tomemos un ejemplo para ilustrar cómo DESeq2 escala las distintas muestras:
Gen
Muestra 1
Muestra 2
Muestra 3
A
0
10
4
B
2
6
12
C
33
55
200
El objetivo es calcular un factor de escala para cada muestra, que tenga en cuenta la profundidad de lectura y la composición de la biblioteca.
Tomar el log\(\_e\) de todos los valores:
Gen
log(Muestra 1)
log(Muestra 2)
log(Muestra 3)
A
-Inf
2.3
1.4
B
0.7
1.8
2.5
C
3.5
4.0
5.3
Promedio de cada fila:
Gen
Media de los valores logarítmicos
A
-Inf
B
1,7
C
4,3
La media de los valores logarítmicos (también conocida como media geométrica) se utiliza aquí porque no se ve afectada fácilmente por valores atípicos (por ejemplo, el gen C con su valor atípico para la Muestra 3).
Filtra los genes cuyo valor es infinito.
Gen
Media de los valores logarítmicos
B
1,7
C
4,3
Aquí filtramos los genes sin recuento de lecturas en al menos 1 muestra, por ejemplo, genes que sólo se transcriben en un tejido como el gen D en el ejemplo anterior. Esto ayuda a centrar los factores de escala en genes transcritos a niveles similares, independientemente de la condición.
Reste el valor logarítmico medio de los recuentos logarítmicos:
Gen
log(Muestra 1)
log(Muestra 2)
log(Muestra 3)
B
-1.0
0.1
0.8
C
-0.8
-0.3
1.0
\[log(\textrm{cuentas del gen X}) - media(\textrm{valores logarítmicos de las cuentas del gen X}) = log(\frac{\textrm{cuentas del gen X}}{\textrm{media del gen X}})\]
Este paso compara la relación entre los recuentos de cada muestra y la media de todas las muestras.
Calcule la mediana de las proporciones de cada muestra:
Gen
log(Muestra 1)
log(Muestra 2)
log(Muestra 3)
B
-1.0
0.1
0.8
C
-0.8
-0.3
1.0
Mediana
-0.9
-0.1
0.9
La mediana se utiliza aquí para evitar que los genes extremos (probablemente los menos frecuentes) influyan demasiado en el valor en una dirección. Ayuda a poner más énfasis en los genes moderadamente expresados.
Calcule el factor de escala tomando la exponencial de las medianas:
Gen
Muestra 1
Muestra 2
Muestra 3
Mediana
-0,9
-0,1
0,9
Factores de escala
0,4
0,9
2,5
Calcular los recuentos normalizados: dividir los recuentos originales por los factores de escala:
DESeq2 también ejecuta el análisis de Expresión Génica Diferencial (GED), que tiene dos tareas básicas:
Estimación de la varianza biológica utilizando las réplicas para cada condición
Estimación de la importancia de las diferencias de expresión entre dos condiciones cualesquiera
Este análisis de expresión se estima a partir de recuentos de lecturas y se intenta corregir la variabilidad en las mediciones utilizando réplicas, que son absolutamente esenciales para obtener resultados precisos. Para su propio análisis, le aconsejamos que utilice al menos 3, pero preferiblemente 5 réplicas biológicas por condición. Es posible tener diferentes números de réplicas por condición.
Una réplica técnica es un experimento que se realiza una vez pero se mide varias veces (por ejemplo, secuenciación múltiple de la misma biblioteca). Una réplica biológica es un experimento realizado (y también medido) varias veces.
En nuestros datos, tenemos 4 réplicas biológicas (aquí llamadas muestras) sin tratamiento y 3 réplicas biológicas con tratamiento (gen Pasilla agotado por RNAi).
Recomendamos combinar las tablas de recuento para diferentes réplicas técnicas (pero no para réplicas biológicas) antes de un análisis de expresión diferencial (véase documentación DESeq2)
A continuación, se pueden incorporar al análisis múltiples factores con varios niveles que describan fuentes de variación conocidas (por ejemplo, tratamiento, tipo de tejido, sexo, lotes), con dos o más niveles que representen las condiciones de cada factor. Tras la normalización, podemos comparar la respuesta de la expresión de cualquier gen a la presencia de distintos niveles de un factor de forma estadísticamente fiable.
En nuestro ejemplo, tenemos muestras con dos factores variables que pueden contribuir a las diferencias en la expresión génica:
Tratamiento (tratado o no tratado)
Tipo de secuenciación (paired-end o single-end)
Aquí, el tratamiento es el factor principal que nos interesa. El tipo de secuenciación es información adicional que conocemos sobre los datos y que podría afectar al análisis. El análisis multifactorial nos permite evaluar el efecto del tratamiento, teniendo también en cuenta el tipo de secuenciación.
Comentario
Le recomendamos que añada todos los factores que crea que pueden afectar a la expresión génica en su experimento. Puede ser el tipo de secuenciación como aquí, pero también puede ser la manipulación (si hay diferentes personas involucradas en la preparación de la librería), otros efectos de lote, etc…
Si sólo tiene uno o dos factores con un número reducido de réplicas biológicas, la configuración básica de DESeq2 es suficiente. En el caso de una configuración experimental compleja con un gran número de réplicas biológicas, las colecciones basadas en etiquetas son apropiadas. Ambos enfoques dan los mismos resultados. El enfoque basado en etiquetas requiere algunos pasos adicionales antes de ejecutar la herramienta DESeq2, pero valdrá la pena cuando se trabaje con una configuración experimental compleja.
Hands-on: Choose Your Own Tutorial
Esto es una sección 'Choose Your Own Tutorial' (CYOT) (también conocida como 'Choose Your Own Analysis' (CYOA)), donde puedes elegir entre varios caminos. Haz click en alguno de los botones debajo para elegir qué quieres seguir en este tutorial
DESeq2 ( Galaxy version 2.11.40.8+galaxy0) con los siguientes parámetros:
“cómo “: Select datasets per level
En “Factor “:
“Specify a factor name, e.g. effects_drug_x or cancer_markers” “: Treatment
En “1: Factor level “:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: treated
En “Count file(s) “: Select all the treated count files (GSM461179, GSM461180, GSM461181)
En “2: Factor level “:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: untreated
En “Count file(s) “: Select all the untreated count files (GSM461176, GSM461177, GSM461178, GSM461182)
param-repeat *“Insertion factor
“Specify a factor name, e.g. effects_drug_x or cancer_markers” “: Sequencing
En “Factor level “:
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: PE
En “Count file(s) “: Select all the paired-end count files (GSM461177, GSM461178, GSM461180, GSM461181)
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: SE
En “Count file(s) “: Select all the single-end count files (GSM461176, GSM461179, GSM461182)
“Files have header? “: Yes
“Choice of input data”: Count data (e.g. from HTSeq-count, featureCounts or StringTie)
En “Advanced options “:
“Use beta priors “: Yes
En “Output options “:
“Output selector “: Generate plots for visualizing the analysis results, Output normalised counts
DESeq2 requiere proporcionar para cada factor, recuentos de muestras en cada categoría. Por lo tanto, utilizaremos etiquetas en nuestra colección de recuentos para seleccionar fácilmente todas las muestras que pertenecen a la misma categoría. Para más información sobre formas alternativas de establecer etiquetas de grupo, consulte este tutorial.
Práctica: Añada etiquetas a su colección para cada uno de estos factores
Cree una lista de colección con todos estos recuentos que etiquete como all counts. Nombre cada elemento de forma que sólo contenga el identificador GSM, el tratamiento y la biblioteca, por ejemplo, GSM461176_untreat_single.
Haz clic en galaxy-selectorSeleccionar elementos en la parte superior del panel del historial
Marque todos los conjuntos de datos de su historial que desee incluir
Haga clic en n of N selected y elija Build dataset list
Introduzca un nombre para su colección
Haz clic en Create collection para crear tu colección
Vuelve a hacer clic en el icono de la marca de verificación situado en la parte superior del historial
Extract element identifiers ( Galaxy version 0.0.2) con los siguientes parámetros:
param-collection“Dataset collection “: all counts
Ahora extraeremos de los nombres los factores:
Replace Text in entire line ( Galaxy version 9.3+galaxy1)
param-file“Fichero a procesar “: salida de Extraer identificadores de elementostool
En “Replacement “:
En “1: Replacement “
“Find pattern “: (.*)_(.*)_(.*)
“Replace with “: \1_\2_\3\tgroup:\2\tgroup:\3
Este paso crea 2 columnas adicionales con el tipo de tratamiento y secuenciación que pueden utilizarse con la herramienta Tag elements tool
Cambie el tipo de datos a tabular
Selecciona sobre el galaxy-pencilicono del lápiz para editar los atributos del conjunto de datos
Selecciona en la pestaña galaxy-chart-select-dataDatatypes en la parte superior del panel central
Selecciona tabular
Da clic en el botón Change datatype
Etiquetar elementos
param-collection“Input Collection “: all counts
param-file“Tag collection elements according to this file “: salida de Replace Texttool
Inspeccionar la nueva colección
Puede que no lo vea a primera vista, ya que los nombres son los mismos. Sin embargo, si hace clic en uno de ellos y en galaxy-tagsEdit dataset tags, deberías ver 2 etiquetas que empiezan por ‘group:’. Esta palabra clave permitirá utilizar estas etiquetas en DESeq2.
DESeq2 ( Galaxy version 2.11.40.8+galaxy0) con los siguientes parámetros:
“how “: Select group tags corresponding to levels
param-collection“Count file(s) collection “: salida de Elementos de etiquetatool
En “Factor “:
param-repeat *“Insertion factor
“Specify a factor name, e.g. effects_drug_x or cancer_markers” “: Treatment
En “Factor level “:
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: treated
“Select groups that correspond to this factor level “: Tags: treat
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: untreated
“Select groups that correspond to this factor level “: Tags: untreat
param-repeat *“Insertion factor
“Specify a factor name, e.g. effects_drug_x or cancer_markers” “: Sequencing
En “Factor level “:
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: PE
“Select groups that correspond to this factor level “: Tags: paired
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: SE
“Select groups that correspond to this factor level “: Tags: single
“Files have header? “: Yes
“Choice of input data”: Count data (e.g. from HTSeq-count, featureCounts or StringTie)
En “Advanced options “:
“Use beta priors “: Yes
En “Output options “:
“Output selector “: Generate plots for visualizing the analysis results, Output normalised counts
DESeq2 requiere proporcionar para cada factor, recuentos de muestras en cada categoría. Por lo tanto, utilizaremos patrones en el nombre de nuestras muestras para seleccionar fácilmente todas las muestras que pertenecen a la misma categoría.
Práctica: Generar una colección de cada categoría
Cree una lista de colección con todos estos recuentos que etiquete como all counts. Nombre cada elemento de forma que sólo contenga el identificador GSM, el tratamiento y la biblioteca, por ejemplo, GSM461176_untreat_single.
Haz clic en galaxy-selectorSeleccionar elementos en la parte superior del panel del historial
Marque todos los conjuntos de datos de su historial que desee incluir
Haga clic en n of N selected y elija Build dataset list
Introduzca un nombre para su colección
Haz clic en Create collection para crear tu colección
Vuelve a hacer clic en el icono de la marca de verificación situado en la parte superior del historial
Extract element identifiers ( Galaxy version 0.0.2) con los siguientes parámetros:
param-collection“Dataset collection “: all counts
Ahora dividiremos la colección por tratamiento. Tenemos que encontrar un patrón que sólo esté presente en una de las 2 categorías. Utilizaremos la palabra untreat:
Search in textfiles ( Galaxy version 9.3+galaxy1) (grep) con los siguientes parámetros:
“Select lines from “: Extract element identifiers on data XXX (salida de Extract element identifierstool)
“that “: Match
“Regular expression “: untreat
Filter collecion con los siguientes parámetros:
“Input collection “: all counts
“How should the elements to remove be determined”: Remove if identifiers are ABSENT from file
“Filter out identifiers absent from “: Search in textfiles on data XXX (salida de Búsqueda en archivos de textotool)
Renombrar ambas colecciones untreated (la colección filtrada) y treated (la colección descartada).
Repetiremos el mismo proceso utilizando single
Search in textfiles ( Galaxy version 9.3+galaxy1) (grep) con los siguientes parámetros:
“Select lines from “: Extract element identifiers on data XXX (salida de Extract element identifierstool)
“that “: Match
“Regular expression “: single
Filter collecion con los siguientes parámetros:
“Input collection “: all counts
“How should the elements to remove be determined”: Remove if identifiers are ABSENT from file
“Filter out identifiers absent from “: Search in textfiles on data XXX (salida de Búsqueda en archivos de textotool)
Renombrar ambas colecciones single (la colección filtrada) y paired (la colección descartada).
DESeq2 ( Galaxy version 2.11.40.8+galaxy0) con los siguientes parámetros:
“cómo “: Select datasets per level
En “Factor “:
“Specify a factor name, e.g. effects_drug_x or cancer_markers” “: Treatment
En “1: Factor level “:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: treated
param-collection“Contar fichero(s) “: Seleccione la colección treated
En “2: Factor level “:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: untreated
param-collection“Contar fichero(s) “: Seleccione la colección untreated
param-repeat *“Insertion factor
“Specify a factor name, e.g. effects_drug_x or cancer_markers” “: Sequencing
En “Factor level “:
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: PE
param-collection“Contar fichero(s) “: Seleccione la colección paired
param-repeat“Insert Factor level “
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’: SE
param-collection“Contar fichero(s) “: Seleccione la colección single
“Files have header? “: Yes
“Choice of input data”: Count data (e.g. from HTSeq-count, featureCounts or StringTie)
En “Advanced options “:
“Use beta priors “: Yes
En “Output options “:
“Output selector “: Generate plots for visualizing the analysis results, Output normalised counts
DESeq2 generó 3 resultados:
Una tabla con los recuentos normalizados para cada gen (filas) en las muestras (columnas)
Resumen gráfico de los resultados, útil para evaluar la calidad del experimento:
Un gráfico de las 2 primeras dimensiones de un análisis de componentes principales (PCA), ejecutado sobre los recuentos normalizados de las muestras
Imaginemos que tenemos algunas botellas de cerveza sobre la mesa. Podemos describir cada cerveza por su color, su espuma, su graduación, etcétera. Podemos componer toda una lista de características diferentes de cada cerveza en una cervecería. Pero muchas de ellas medirán propiedades relacionadas y, por tanto, serán redundantes. Si es así, deberíamos poder resumir cada cerveza con menos características. Esto es lo que hace el ACP o análisis de componentes principales.
Con el ACP, no nos limitamos a seleccionar algunas características interesantes y descartar las demás. En su lugar, construimos algunas características nuevas que resumen bien nuestra lista de cervezas. Estas nuevas características se construyen a partir de las anteriores. Por ejemplo, se puede calcular una nueva característica, como el tamaño de la espuma menos el pH de la cerveza. Se trata de combinaciones lineales.
De hecho, PCA encuentra las mejores características posibles, las que resumen la lista de cervezas. Estas características pueden utilizarse para encontrar similitudes entre las cervezas y agruparlas.
Volviendo a los recuentos de lecturas, el PCA se ejecuta en los recuentos normalizados de todas las muestras. Aquí, nos gustaría describir las muestras basándonos en la expresión de los genes. Así que las características son el número de lecturas mapeadas en cada gen. Las utilizamos, así como combinaciones lineales de las mismas, para representar las muestras y sus similitudes.
Muestra las muestras en el plano 2D abarcado por sus dos primeros componentes principales. Cada réplica se representa como un punto de datos individual. Este tipo de gráfico es útil para visualizar el efecto global de las covariables experimentales y los efectos de lote.
Figura 40: Diagrama de componentes principales de las muestras
¿Qué separa la primera dimensión (PC1)?
¿Y la segunda dimensión (PC2)?
¿Qué podemos concluir sobre el diseño DESeq (factores, niveles) que elegimos?
La primera dimensión es separar las muestras tratadas de las no tratadas.
La segunda dimensión consiste en separar los conjuntos de datos de extremo único de los conjuntos de datos de extremo pareado.
Los conjuntos de datos se agrupan siguiendo los niveles de los dos factores. No parece haber ningún efecto oculto en los datos. Si hay una variación no deseada presente en los datos (por ejemplo, efectos de lote), siempre se recomienda corregirla, lo que puede lograrse en DESeq2 incluyendo en el diseño cualquier variable de lote conocida.
Heatmap de la matriz de distancias muestra-muestra (con agrupación) basada en los recuentos normalizados.
El mapa de calor ofrece una visión general de las similitudes y disimilitudes entre las muestras: el color representa la distancia entre las muestras. El azul oscuro significa una distancia más corta, es decir, muestras más cercanas dados los recuentos normalizados.
Figura 41: Mapa térmico de las distancias entre muestras
¿Cómo se agrupan las muestras?
En primer lugar, se agrupan según el tratamiento (primer factor) y, en segundo lugar, según el tipo de secuenciación (segundo factor), como en el gráfico PCA.
Estimaciones de dispersión: estimaciones por genes (negro), los valores ajustados (rojo) y las estimaciones finales máximas a posteriori utilizadas en las pruebas (azul)
Este gráfico de dispersión es típico, con las estimaciones finales reducidas desde las estimaciones por genes hacia las estimaciones ajustadas. Algunas estimaciones por genes se marcan como valores atípicos y no se reducen hacia el valor ajustado. La cantidad de contracción puede ser mayor o menor de lo que se ve aquí, dependiendo del tamaño de la muestra, el número de coeficientes, la media de la fila y la variabilidad de las estimaciones por genes.
Histograma de valores p para los genes en la comparación entre los 2 niveles del 1er factor
Muestra la vista global de la relación entre el cambio de expresión de las condiciones (log ratios, M), la fuerza de expresión media de los genes (media media, A) y la capacidad del algoritmo para detectar la expresión génica diferencial. Los genes que superaron el umbral de significación (valor p ajustado < 0,1) están coloreados en rojo.
Un archivo de resumen con los siguientes valores para cada gen:
Identificadores de genes
Recuentos medios normalizados, promediados en todas las muestras de ambas condiciones
Doble cambio en log2 (logaritmo base 2)
Los cambios de pliegues log2 se basan en el factor primario de nivel 1 frente al factor de nivel 2, por lo que el orden de entrada de los niveles del factor es importante. Aquí, DESeq2 calcula los cambios de pliegues de las muestras “tratadas” frente a las “no tratadas” a partir del primer factor “Tratamiento”, es decir los valores corresponden a la regulación al alza o a la baja de los genes en las muestras tratadas.
Estimación del error estándar para la estimación del cambio de pliegue log2
Valor p para la significación estadística de este cambio
valor p ajustado para pruebas múltiples con el procedimiento Benjamini-Hochberg, que controla la tasa de falsos descubrimientos (FDR)
El valor p es una medida utilizada a menudo para determinar si una observación concreta posee o no significación estadística. En sentido estricto, el valor p es la probabilidad de que los datos hayan podido surgir al azar, suponiendo que la hipótesis nula sea correcta. En el caso concreto de RNA-Seq, la hipótesis nula es que no hay expresión génica diferencial. Por lo tanto, un valor p de 0,13 para un gen concreto indica que, para ese gen, suponiendo que no se exprese de forma diferencial, hay un 13% de posibilidades de que cualquier expresión diferencial aparente pueda producirse simplemente por una variación aleatoria en los datos experimentales.
el 13% sigue siendo bastante elevado, por lo que no podemos estar seguros de que se esté produciendo una expresión génica diferencial. La forma más común en que los científicos utilizan los valores p es establecer un umbral (normalmente 0,05, a veces otros valores como 0,01) y rechazar la hipótesis nula sólo para valores p por debajo de este valor. Así, para los genes con valores p inferiores a 0,05, podemos afirmar con seguridad que la expresión génica diferencial desempeña un papel. Hay que tener en cuenta que cualquier umbral de este tipo es arbitrario y que no hay ninguna diferencia significativa entre un valor p de 0,049 y 0,051, aunque sólo rechacemos la hipótesis nula en el primer caso.
Desgraciadamente, los p-valores a menudo se utilizan mal en la investigación científica, hasta el punto de que Wikipedia ofrece un artículo dedicado sobre el tema. Véase también este artículo (dirigido a un público general, no científico).
Para más información sobre DESeq2 y sus resultados, puede consultar la documentación de DESeq2.
Preguntas
¿Se expresa de forma diferencial el gen FBgn0003360 debido al tratamiento? En caso afirmativo, ¿en qué medida?
¿Está el gen Pasilla (ps, FBgn0261552) regulado a la baja por el tratamiento de ARNi?
También podríamos estar hipotéticamente interesados en el efecto de la secuenciación (u otros factores secundarios en otros casos). ¿Cómo conoceríamos los genes expresados diferencialmente debido al tipo de secuenciación?
Nos gustaría analizar la interacción entre el tratamiento y la secuenciación. ¿Cómo podríamos hacerlo?
FBgn0003360 se expresa de forma diferencial debido al tratamiento: tiene un valor p ajustado significativo (\(2,8 \cdot 10^{-171} << 0,05\)). Se expresa menos (- en la columna log2FC) en las muestras tratadas en comparación con las no tratadas, en un factor ~8 (\(2^{log2FC} = 2^{2.99542318410271}\)).
Puede comprobar manualmente si hay FBgn0261552 en la primera columna o ejecutar Filter data on any column using simple expressions
param-file“Filter “: el DESeq2 result file (salida de DESeq2tool)
“With condition”: c1 == "FBgn0261552"
El cambio de pliegues log2 es negativo, por lo que está regulado a la baja, y el valor p ajustado es inferior a 0,05, por lo que forma parte de los genes con cambios significativos.
DESeq2 en Galaxy devuelve la comparación entre los distintos niveles para el 1er factor, tras la corrección por la variabilidad debida al 2º factor. En nuestro caso actual, tratado contra no tratado para cualquier tipo de secuenciación. Para comparar los tipos de secuenciación, deberíamos ejecutar DESeq2 de nuevo cambiando los factores: el factor 1 (tratamiento) se convierte en el factor 2 y el factor 2 (secuenciación) se convierte en el factor 1.
Para añadir la interacción entre dos factores (por ejemplo, tratado para datos paired-end vs no tratado para single-end), debemos ejecutar DESeq2 otra vez pero con un solo factor con los siguientes 4 niveles:
tratado-PE
no tratado-PE
tratado-SE
sin tratar-SE
Seleccionando “Output all levels vs all levels of primary factor (use when you have >2 levels for primary factor) “ a Yes, podemos comparar treated-PE vs untreated-SE.
Anotación de los resultados de DESeq2
El ID de cada gen es algo así como FBgn0003360, que es un ID de la base de datos correspondiente, aquí Flybase (Thurmond et al. 2018). Estos IDs son únicos, pero a veces preferimos tener los nombres de los genes, incluso si pueden no hacer referencia a un gen único (por ejemplo, duplicado después de la re-anotación). Sin embargo, los nombres de los genes pueden indicar ya una función o ayudar a buscar los candidatos deseados. También nos gustaría mostrar la localización de estos genes dentro del genoma. Podemos extraer dicha información del archivo de anotación que hemos utilizado para el mapeo y el recuento.
Práctica: Anotación de los resultados de DESeq2
Importar la anotación de genes Ensembl para Drosophila melanogaster (Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz) del historial anterior, o de la biblioteca de Datos Compartidos o de Zenodo:
Annotate DESeq2/DEXSeq output tables ( Galaxy version 1.1.0) with:
param-file“Tabular output of DESeq2/edgeR/limma/DEXSeq “: the DESeq2 result file (output of DESeq2tool)
“Input file type “: DESeq2/edgeR/limma
param-file“Reference annotation in GFF/GTF format “: imported gtf Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
La salida generada es una extensión del archivo anterior:
Identificadores de genes
Recuentos medios normalizados de todas las muestras
Cambio de pliegue Log2
Estimación del error estándar para la estimación del cambio de pliegue log2
Estadística de Wald
Valor p para el estadístico de Wald
valor p ajustado para pruebas múltiples con el procedimiento Benjamini-Hochberg para la estadística Wald
Cromosoma
Inicio
Fin
Hebra
Característica
Nombre del gen
Preguntas
¿Dónde se encuentra el gen más sobreexpresado?
¿Cuál es el nombre del gen?
¿Dónde se encuentra el gen Pasilla (FBgn0261552)?
FBgn0025111 (el gen mejor clasificado con el valor log2FC positivo más alto) se encuentra en el reverso del cromosoma X, entre 10.778.953 pb y 10.786.907 pb.
De la tabla, obtenemos el símbolo del gen: Ant2. Después de algunas búsquedas en las bases de datos biológicas en línea, encontramos que Ant2 corresponde a la adenina nucleótido translocasa 2.
El gen Pasilla se encuentra en la cadena anterior del cromosoma 3R, entre 9.417.939 pb y 9.455.500 pb.
La tabla de anotaciones no contiene nombres de columnas, lo que dificulta su lectura. Nos gustaría añadirlos antes de continuar.
Práctica: Añadir nombres de columnas
Cree un nuevo archivo (header) a partir de lo siguiente (línea de encabezado de la salida DESeq2)
GeneID Base mean log2(FC) StdErr Wald-Stats P-value P-adj Chromosome Start End Strand Feature Gene name
Pulse galaxy-uploadCargar Datos en la parte superior del panel de herramientas
Seleccione galaxy-wf-editPegar/Obtener Datos en la parte inferior
Pegue el contenido del archivo en el campo de texto
Cambie el nombre del conjunto de datos de “New File” a header * Cambie Type de “Auto-detect” a tabular* Pulse Iniciar y Cerrar la ventana
Concatenate multiple datasets or collections para añadir esta línea de cabecera a la salida Annotate:
param-file“Concatenate Dataset “: el conjunto de datos header
“Dataset “
Haga clic en param-repeat“Insertar conjunto de datos “
param-file“select “: salida de Annotatetool
Cambie el nombre de la salida a Annotated DESeq2 results
Extracción y anotación de genes expresados diferencialmente
Ahora queremos extraer los genes más expresados diferencialmente debido al tratamiento con un cambio de pliegue > 2 (o < 1/2).
Práctica: Extraer los genes más expresados diferencialmente
Filter data on any column using simple expressions para extraer los genes con un cambio significativo en la expresión génica (valor p ajustado inferior a 0,05) entre las muestras tratadas y no tratadas:
param-file“Filter “: Annotated DESeq2 results
“With following condition”: c7<0.05
“Number of header lines to skip “: 1
Renombrar la salida Genes with significant adj p-value.
Preguntas
¿Cuántos genes presentan un cambio significativo en la expresión génica entre estas condiciones?
Obtenemos 966 (967 líneas incluyendo una cabecera) genes (4,04%) con un cambio significativo en la expresión génica entre las muestras tratadas y no tratadas.
Comentario
El archivo con los resultados filtrados de forma independiente puede utilizarse para análisis posteriores, ya que excluye genes con sólo unos pocos recuentos de lecturas, ya que estos genes no se considerarán expresados de forma diferencial significativa.
Ahora seleccionaremos sólo los genes con un cambio de pliegue (FC) > 2 o FC < 0,5. Tenga en cuenta que el archivo de salida de DESeq2 contiene \(log\_{2} FC\), en lugar del propio FC, por lo que filtraremos por \(abs(log\_{2} FC) > 1\) (lo que implica FC > 2 o FC < 0,5).
Filter data on any column using simple expressions para extraer genes con un \(abs(log\_{2} FC) > 1\):
param-file“Filter “: Genes with significant adj p-value
“With following condition”: abs(c3)>1
“Number of header lines to skip “: 1
Renombrar la salida Genes with significant adj p-value & abs(log2(FC)) > 1.
Preguntas
¿Cuántos genes se han conservado?
¿Se puede encontrar el gen Pasilla (ps, FBgn0261552) en esta tabla?
Obtenemos 113 genes (114 líneas incluida la cabecera), es decir, el 11,79% de los genes con expresión diferencial significativa.
El gen Pasilla puede encontrarse con una búsqueda rápida (o incluso utilizando Filter data on any column using simple expressions )
Ahora tenemos una tabla con 113 líneas y una cabecera correspondiente a los genes más expresados diferencialmente. Para cada gen, tenemos su ID, sus recuentos medios normalizados (promediados sobre todas las muestras de ambas condiciones), su \(log\_{2} FC\) y otra información que incluye el nombre y la posición del gen.
Visualización de la expresión de los genes expresados diferencialmente
Podríamos trazar el \(log\_{2} FC\) para los genes extraídos, pero aquí nos gustaría ver un mapa de calor de la expresión de estos genes en las diferentes muestras. Así que tenemos que extraer los recuentos normalizados para estos genes.
Procedemos en varios pasos:
Extraiga y trace los recuentos normalizados de estos genes para cada muestra con un mapa de calor, utilizando el archivo de recuentos normalizados generado por DESeq2
Calcular, extraer y representar gráficamente la puntuación Z de los recuentos normalizados
Comentario: Tutoriales avanzados sobre visualización
En este tutorial, explicaremos rápidamente algunas posibilidades de visualización. Para más detalles, eche un vistazo a los tutoriales adicionales sobre visualización de resultados de RNA-Seq:
Para extraer los recuentos normalizados de los genes interesantes, unimos la tabla de recuentos normalizados generada por DESeq2 con la tabla que acabamos de generar. Conservaremos entonces sólo las columnas correspondientes a los recuentos normalizados.
Práctica: Extraer los recuentos normalizados de los genes más diferencialmente expresados
Join two Datasets side by side on a specified field con los siguientes parámetros:
param-file“Join “: el archivo Normalized counts (salida de DESeq2tool)
“Keep lines of first input that do not join with second input “: No
“Keep header files “: Yes
El archivo generado tiene más columnas de las que necesitamos para el mapa de calor: recuentos medios normalizados, \(log\_{2} FC\) y otra información de anotación. Tenemos que eliminar las columnas adicionales.
Cut columnas de una tabla con los siguientes parámetros para extraer las columnas con los IDs de los genes y los recuentos normalizados:
“Cut columns “: c1-c8
“Delimited by “: Tab
param-file“From “: el conjunto de datos unido (resultado de Join two Datasetstool)
Cambie el nombre de la salida a Normalized counts for the most differentially expressed genes
Ahora tenemos una tabla con 114 líneas (los 113 genes más expresados diferencialmente y una cabecera) y los recuentos normalizados de estos genes en las 7 muestras.
Práctica: Planifica el mapa de calor de los recuentos normalizados de estos genes para las muestras
heatmap2 ( Galaxy version 3.1.3.1+galaxy0) para trazar el mapa de calor:
param-file“Input should have column headers “: Normalized counts for the most differentially expressed genes
“Data transformation “: Log2(value+1) transform my data
“Enable data clustering “: Yes
“Labeling columns and rows “: Label columns and not rows
“Type of colormap to use “: Gradient with 2 colors
Figura 42: Recuentos normalizados de los genes más expresados diferencialmente
Preguntas
¿Qué representa el eje X del mapa de calor? ¿Y el eje Y?
¿Observa algo en la agrupación de las muestras y los genes?
¿Qué cambia si regenera el mapa de calor, esta vez seleccionando Plot the data as it is en “Data transformation “?
¿Por qué no podemos utilizar Log2(value) transform my data en “Data transformation “?
¿Cómo podría generar un mapa térmico de recuentos normalizados para todos los genes regulados al alza con un cambio de pliegue > 2?
El eje X muestra las 7 muestras, junto con un dendrograma que representa la similitud entre sus niveles de expresión génica. El eje Y muestra los 113 genes expresados diferencialmente, igualmente con un dendrograma que representa la similitud entre los niveles de expresión génica.
Las muestras se agrupan por tratamiento.
La escala cambia y sólo vemos unos pocos genes.
Porque la expresión normalizada del gen FBgn0013688 en GSM461180_treat_paired está en 0.
Extraer los genes con \(log\_{2} FC\) > 1 (filtro para genes con c3>1 en el resumen de los genes expresados diferencialmente) y ejecute heatmap2tool en la tabla generada.
Visualización de la puntuación Z
Para comparar la expresión génica entre muestras, también podríamos utilizar la puntuación Z, que a menudo se representa en las publicaciones.
La puntuación Z da el número de desviaciones estándar que un valor se aleja de la media de todos los valores del mismo grupo, aquí el mismo gen. Una puntuación Z de -2 para el gen X en la muestra A significa que este valor es 2 desviaciones estándar inferior a la media de los valores del gen X en todas las muestras (A, B, C, etc.).
La puntuación Z \(z\_{i,j}\) para un gen \(i\) en una muestra \(j\) dado el recuento normalizado \(x\_{i,j}\) se calcula como \(z\_{i,j} = \frac{x\_{i,j}- \overline{x\_i}}{s\_i}\) con \(\overline{x\_i}\) la media y \(s\_i\) la desviación estándar de los recuentos normalizados para el gen \(i\) en todas las muestras.
A menudo necesitamos la puntuación Z para algunas visualizaciones. Para calcular la puntuación Z, dividimos el proceso en 2 pasos:
Reste cada valor por la media de los valores de la fila (es decir, \(x\_{i,j}- \overline{x\_i}\)) utilizando la tabla de recuento normalizado
Dividir los valores anteriores por la desviación estándar de los valores de la fila, utilizando 2 tablas (los recuentos normalizados y la tabla calculada en el paso anterior)
Práctica: Calcular la puntuación Z de todos los genes
Table Compute ( Galaxy version 1.2.4+galaxy0) con los siguientes parámetros para > restar primero los valores medios por fila
“Input Single or Multiple Tables “: Single Table
param-file“Table “: Normalized counts file on data ... and others (salida de DESeq2tool)
“Type of table operation “: Perform a full table operation
“Operation “: Custom
“Custom expression on ‘table’, along ‘axis’ (0 or 1) “: table.sub(table.mean(1), 0)
La expresión table.mean(1) calcula la media de cada fila (aquí los genes) y table.sub(table.mean(1), 0) resta a cada valor la media de la fila (calculada con table.mean(1))
Table Compute ( Galaxy version 1.2.4+galaxy0) con los siguientes parámetros:
“Input Single or Multiple Tables “: Multiple Table
Haga clic en param-repeat“Insert tables “
En “1: Tablas “:
param-file“Table “: Normalized counts file on data ... and others (salida de DESeq2tool)
Haga clic en param-repeat“Insert tables “
En “2: Tablas “:
param-file“Table “: salida del primer Table Computetool
“Custom expression on ‘tableN: table2.div(table1.std(1),0)
La expresión table1.std(1) calcula las desviaciones estándar de cada fila de la 1ª tabla (recuentos normalizados) y table2.div divide los valores de la 2ª tabla (calculados previamente) por estas desviaciones estándar.
Cambie el nombre de la salida a Z-scores
Inspeccione el archivo de salida
Ahora tenemos una tabla con la puntuación Z de todos los genes de las 7 muestras.
Preguntas
¿Cuál es el rango para la puntuación Z?
¿Por qué algunas filas están vacías?
¿Qué podemos decir de las puntuaciones Z de los genes expresados diferencialmente (por ejemplo, FBgn0037223)?
¿Podemos utilizar la puntuación Z para estimar la intensidad de la expresión diferencial de un gen?
La puntuación Z oscila entre -3 desviaciones estándar y +3 desviaciones estándar. Puede situarse en una curva de distribución normal: -3 es el extremo izquierdo de la curva de distribución normal y +3 el extremo derecho de la curva de distribución normal
Si todos los recuentos son idénticos (normalmente a 0), la desviación estándar es 0, la puntuación Z no puede calcularse para estos genes.
Cuando un gen se expresa diferencialmente entre dos grupos (aquí tratado y no tratado), las puntuaciones Z para este gen serán (mayoritariamente) positivas para las muestras de un grupo y (mayoritariamente) negativas para las muestras del otro grupo.
La puntuación Z es una relación señal-ruido. Las puntuaciones Z absolutas grandes, es decir, los valores positivos o negativos grandes, no son una estimación directa del efecto, es decir, de la fuerza de la expresión diferencial. Una misma puntuación Z grande puede tener diferentes significados, dependiendo del ruido:
con mucho ruido: un efecto muy grande
con algo de ruido: un efecto bastante grande
con poco ruido: un efecto más bien pequeño
casi sin ruido: un efecto minúsculo
El problema es que aquí el “ruido” no es sólo ruido de la medida. También puede estar relacionado con el “rigor” del control de la regulación de los genes. Los genes no estrechamente controlados, es decir, cuya expresión puede variar en un amplio rango a lo largo de las muestras, pueden ser inducidos o reprimidos considerablemente. Su puntuación Z absoluta será pequeña, ya que las variaciones entre muestras son grandes. Por el contrario, los genes que están estrechamente controlados pueden tener sólo cambios muy pequeños en su expresión, sin ningún impacto biológico. La puntuación Z absoluta será grande para estos genes.
Ahora nos gustaría trazar un mapa de calor para las puntuaciones Z:
Figura 43: Puntuaciones Z de los genes con mayor expresión diferencial
Práctica: Planificar la puntuación Z de los genes más expresados diferencialmente
heatmap2 ( Galaxy version 3.1.3.1+galaxy0) para trazar el mapa de calor:
param-file“Input should have column headers “: Normalized counts for the most differentially expressed genes
“Data transformation “: Plot the data as it is
“Compute z-scores prior to clustering “: Compute on rows
“Enable data clustering “: Yes
“Labeling columns and rows “: Label columns and not rows
“Type of colormap to use “: Gradient with 3 colors
Análisis de enriquecimiento funcional de los genes DE
Hemos extraído los genes que se expresan diferencialmente en las muestras tratadas (con genes PS agotados) en comparación con las muestras no tratadas. Ahora, nos gustaría saber si los genes expresados diferencialmente son transcritos enriquecidos de genes que pertenecen a categorías más comunes o específicas para identificar las funciones biológicas que podrían verse afectadas.
Análisis de ontología génica
El análisis Gene Ontology (GO) se utiliza ampliamente para reducir la complejidad y resaltar los procesos biológicos en los estudios de expresión de todo el genoma. Sin embargo, los métodos estándar dan resultados sesgados en los datos de RNA-Seq debido a la sobredetección de la expresión diferencial para transcritos largos y altamente expresados.
goseq (Young et al. 2010) proporciona métodos para realizar análisis GO de datos RNA-Seq teniendo en cuenta el sesgo de longitud. goseq también podría aplicarse a otros análisis basados en categorías de datos RNA-Seq, como el análisis de vías KEGG, como se discute en una sección posterior.
goseq necesita 2 archivos de entrada:
Un archivo tabular con los genes expresados diferencialmente de todos los genes analizados en el experimento RNA-Seq con 2 columnas:
los Gene IDs (únicos dentro del archivo), en mayúsculas
un booleano que indica si el gen se expresa de forma diferencial o no (True si se expresa de forma diferencial o False si no)
Un archivo con información sobre la longitud de un gen para corregir el posible sesgo de longitud en genes expresados diferencialmente
Práctica: Preparar el primer conjunto de datos para goseq
Compute ( Galaxy version 2.0) en filas con los siguientes parámetros:
param-file“Input file”: el DESeq2 result file (salida de DESeq2tool)
En “Expresiones”:
param-text“Add expression”: bool(float(c7)<0.05)
param-select“Mode of the operation?”: Append
En “Tratamiento de errores”:
param-toggle“Autodetect column types”: No
param-select“If an expression cannot be computed for a row”: Fill in a replacement value
param-select“Replacement value”: False
Cut columnas de una tabla con los siguientes parámetros:
“Cut columns”: c1,c8
“Delimited by”: Tab
param-file“From”: la salida de Computetool
Change Case con
param-file“From”: la salida del Cut anterior tool
“Change case of columns”: c1
“Delimited by”: Tab
“To”: Upper case
Cambie el nombre de la salida a Gene IDs and differential expression
Acabamos de generar la primera entrada para goseq. Como segunda entrada para goseq necesitamos las longitudes de los genes. Podemos usar aquí las longitudes de los genes generadas por featureCounts o Gene length and GC content y formatear los IDs de los genes.
Práctica: Preparar el archivo de longitud del gen
Copy the feature length collection previously generated by featureCountstool into this history
Hay 3 formas de copiar conjuntos de datos entre historiales
Del historial original
Haga clic en el icono galaxy-gear que se encuentra en la parte superior de la lista de conjuntos de datos en el panel de historial
Haga clic en Copiar conjuntos de datos
Seleccione los archivos deseados
Dé un nombre relevante al “Nuevo historial”
Validar por ‘Copiar elementos del historial’
Haga clic en el nombre de la nueva historia en el cuadro verde que acaba de aparecer para cambiar a esta historia
Uso del galaxy-columnsMostrar Historias lado a lado
Haga clic en la flecha desplegable galaxy-dropdown arriba a la derecha del panel de historial (Opciones de historial)
Haga clic en galaxy-columnsMostrar Historias lado a lado
Si el historial de destino no está presente
Haga clic en “Seleccionar historiales
Haga clic en el historial de destino
Validar por ‘Cambiar seleccionado’
Arrastre el conjunto de datos a copiar desde su historial original
Colócalo en el historial de destino
Del historial de destino
Haga clic en Usuario en la barra superior
Haga clic en Conjuntos de datos
Buscar el conjunto de datos a copiar
Haga clic en su nombre
Haga clic en Copiar al historial actual
Extraer conjunto de datos con:
param-collection“Input List “: featureCounts on collection N: Feature lengths
“¿Cómo debe seleccionarse un conjunto de datos?: The first dataset
Copy the output of Gene length and GC contenttool (Gene length) into this history
Hay 3 formas de copiar conjuntos de datos entre historiales
Del historial original
Haga clic en el icono galaxy-gear que se encuentra en la parte superior de la lista de conjuntos de datos en el panel de historial
Haga clic en Copiar conjuntos de datos
Seleccione los archivos deseados
Dé un nombre relevante al “Nuevo historial”
Validar por ‘Copiar elementos del historial’
Haga clic en el nombre de la nueva historia en el cuadro verde que acaba de aparecer para cambiar a esta historia
Uso del galaxy-columnsMostrar Historias lado a lado
Haga clic en la flecha desplegable galaxy-dropdown arriba a la derecha del panel de historial (Opciones de historial)
Haga clic en galaxy-columnsMostrar Historias lado a lado
Si el historial de destino no está presente
Haga clic en “Seleccionar historiales
Haga clic en el historial de destino
Validar por ‘Cambiar seleccionado’
Arrastre el conjunto de datos a copiar desde su historial original
Colócalo en el historial de destino
Del historial de destino
Haga clic en Usuario en la barra superior
Haga clic en Conjuntos de datos
Buscar el conjunto de datos a copiar
Haga clic en su nombre
Haga clic en Copiar al historial actual
Change Case con los siguientes parámetros:
param-file“De “: GSM461177_untreat_paired (salida de Extract Datasettool)Gene length
“Change case of columns “: c1
“Delimited by”: Tab
“To “: Upper case
Cambie el nombre de la salida a Gene IDs and length
Ya tenemos los dos archivos de entrada necesarios para goseq.
Práctica: Realizar análisis GO
goseq ( Galaxy version 1.50.0+galaxy0) with
“Differentially expressed genes file “: Gene IDs and differential expression
“Gene lengths file “: Gene IDs and length
“Gene categories “: Get categories
“Select a genome to use “: Fruit fly (dm6)
“Select Gene ID format “: Ensembl Gene ID
“Select one or more categories “: GO: Cellular Component, GO: Biological Process, GO: Molecular Function
En “Output options “
“Output Top GO terms plot? “: Yes
“Extract the DE genes for the categories (GO/KEGG terms)? “: Yes
goseq genera con estos parámetros 3 salidas:
Una tabla (Ranked category list - Wallenius method) con las siguientes columnas para cada término GO:
category: Categoría GO
over_rep_pval: valor p para la sobrerrepresentación del término en los genes expresados diferencialmente
under_rep_pval: valor p para la infrarrepresentación del término en los genes expresados diferencialmente
numDEInCat: número de genes expresados diferencialmente en esta categoría
numInCat: número de genes en esta categoría
term: detalle del término
ontology: MF (Función Molecular - actividades moleculares de los productos génicos), CC (Componente Celular - donde los productos génicos son activos), BP (Proceso Biológico - vías y procesos más amplios compuestos por las actividades de múltiples productos génicos)
p.adjust.over_represented: valor p para la sobrerrepresentación del término en los genes con expresión diferencial, ajustado para pruebas múltiples con el procedimiento Benjamini-Hochberg
p.adjust.under_represented: valor p para la infrarrepresentación del término en los genes con expresión diferencial, ajustado para pruebas múltiples con el procedimiento Benjamini-Hochberg
Para identificar categorías significativamente enriquecidas/no enriquecidas por debajo de algún valor p de corte, es necesario utilizar el valor p ajustado.
Preguntas
¿Cuántos términos GO están sobrerrepresentados con un valor P ajustado < 0,05? ¿Cuántos están infrarrepresentados?
¿Cómo se dividen los términos GO sobrerrepresentados en MF, CC y BP? ¿Y para los términos GO infrarrepresentados?
60 términos GO (0,50%) están sobrerrepresentados y 7 (0,07%) infrarrepresentados.
Filter data on any column using simple expressions on c8 (adjusted p-value for over-represented GO terms) and c9 (adjusted p-value for under-represented GO terms)
Para sobre-representados, 50 BP, 5 CC y 5 MF y para sub-representados, 5 BP, 2 CC y 0 MF
Agrupar datos en la columna 7 (categoría) y recuento en la columna 1 (IDs)
Un gráfico con los 10 términos GO más sobrerrepresentados
Preguntas
¿Qué es el eje x? ¿Cómo se calcula?
El eje x es el porcentaje de genes de la categoría que se han identificado como expresados diferencialmente: \(100 \times \frac{numDEInCat}{numInCat}\)
Una tabla con los genes expresados diferencialmente (de la lista que proporcionamos) asociados a los términos GO (DE genes for categories (GO/KEGG terms))
Comentario: Tutorial avanzado sobre análisis de enriquecimiento
En este tutorial, hemos cubierto el análisis de enriquecimiento GO con goseq. Para aprender otros métodos y herramientas para el análisis de enriquecimiento de conjuntos de genes, por favor eche un vistazo al tutorial “RNA-Seq genes to pathways”.
Análisis de rutas KEGG
goseq también puede utilizarse para identificar vías KEGG interesantes. La base de datos de vías KEGG es una colección de mapas de vías que representan el conocimiento actual de las redes de interacciones, reacciones y relaciones moleculares. Un mapa puede integrar muchas entidades, incluidos genes, proteínas, ARN, compuestos químicos, glicanos y reacciones químicas, así como genes de enfermedades y dianas farmacológicas.
Por ejemplo, la ruta dme00010 representa el proceso de glucólisis (conversión de glucosa en piruvato con generación de pequeñas cantidades de ATP y NADH) para Drosophila melanogaster:
Práctica: Realizar análisis de vías KEGG
goseq ( Galaxy version 1.50.0+galaxy0) with
“Differentially expressed genes file “: Gene IDs and differential expression
“Gene lengths file “: Gene IDs and length
“Gene categories “: Get categories
“Select a genome to use “: Fruit fly (dm6)
“Select Gene ID format “: Ensembl Gene ID
“Select one or more categories “: KEGG
En “Output options “
“Output Top GO terms plot? “: No
“Extract the DE genes for the categories (GO/KEGG terms)? “: Yes
goseq genera con estos parámetros 2 salidas:
Una tabla grande con los términos KEGG y algunas estadísticas
Preguntas
¿Cuántos términos de vías KEGG se han identificado?
¿Cuántos términos de vías KEGG están sobrerrepresentados con un valor P ajustado < 0,05?
¿Cuáles son los términos de vías KEGG sobrerrepresentados?
¿Cuántos términos de vías KEGG están infrarrepresentados con un valor P ajustado < 0,05?
El archivo tiene 128 líneas, incluida la cabecera, por lo que se han identificado 127 vías KEGG.
2 vías KEGG (2,34%) están sobrerrepresentadas, utilizando Filtrar datos en cualquier columna utilizando expresiones simples en c6 (valor p ajustado para vías KEGG sobrerrepresentadas)
Las dos vías KEGG sobrerrepresentadas son 01100 y 00010. Buscándolas en la base de datos KEGG, podemos encontrar más información sobre estas vías: 01100 corresponde a todas las vías metabólicas y 00010 a la vía de la Glucólisis / Gluconeogénesis.
Ninguna vía KEGG está infrarrepresentada, utilizando Filtrar datos en cualquier columna utilizando expresiones simples en c7 (valor p ajustado para vías KEGG infrarrepresentadas)
Una tabla con los genes expresados diferencialmente (de la lista que proporcionamos) asociados a las vías KEGG (DE genes for categories (GO/KEGG terms))
Podríamos investigar qué genes están implicados en qué rutas consultando el segundo archivo generado por goseq. Sin embargo, esto puede ser engorroso y nos gustaría ver las rutas como se representa en la imagen anterior. Pathview (Luo and Brouwer 2013) puede ayudar a generar automáticamente imágenes similares a la anterior, a la vez que añade información extra sobre los genes (por ejemplo, expresión) en nuestro estudio.
Esta herramienta necesita 2 entradas principales:
ID de la(s) vía(s) a trazar, ya sea como un único ID o como un archivo con una columna con los ID de la vía
Un archivo tabular con los genes del experimento RNA-Seq con 2 (o más) columnas:
los ID de los genes (únicos dentro del archivo)
información sobre los genes
Puede ser, por ejemplo, un valor p o un cambio de pliegue. Esta información se añadirá al trazado de la ruta: el nodo del gen correspondiente se coloreará con el valor. Si hay diferentes columnas, las diferentes informaciones se trazarán una al lado de la otra en el nodo.
Aquí nos gustaría visualizar las 2 vías KEGG: la sobrerrepresentada 00010 (Glucólisis / Gluconeogénesis) y la más infrarrepresentada (aunque no significativamente) 03040 (Spliceosoma). Nos gustaría que los nodos de genes fueran coloreados por Log2 Fold Change para los genes diferencialmente expresados debido al tratamiento.
Práctica: Overlay log2FC on KEGG pathway
(Superponer log2FC en la ruta KEGG)
Cut columnas de una tabla con los siguientes parámetros:
“Cut columns “: c1,c3
“Delimited by”: Tab
param-file“De “: Genes with significant adj p-value
Cambie el nombre a Genes with significant adj p-value and their Log2 FC
Extraemos el ID y el Log2 Fold Change de los genes que tienen un valor p ajustado significativo.
Cree un nuevo archivo tabular a partir de lo siguiente (IDs de las rutas a trazar) llamado KEGG pathways to plot
00010
03040
Pathview ( Galaxy version 1.34.0+galaxy0) with
“Number of pathways to plot “: Multiple
param-file“KEGG pathways “: KEGG pathways to plot
“Does the file have header (a first line with column names)? “: No
“Especies a utilizar “: Fly
“Provide a gene data file?: Yes
param-file“Gene data “: Genes with significant adj p-value and their Log2 FC
“Does the file have header (a first line with column names)? “: Yes
“Format for gene data “: Ensembl Gene ID
“Provide a compound data file? “: No
En “Output options “
“Output for pathway “: KEGG native
“Plot on same layer?”: Yes
Pathview genera una colección con la visualización KEGG: un archivo por ruta.
Preguntas
dme00010 Ruta KEGG de Pathview
¿Qué son las cajas de color?
¿Cuál es el código de color?
Los recuadros de color son genes de la vía que se expresan de forma diferencial
Preste atención a que el código de colores es contrario a la intuición: el verde es para valores inferiores a 0, es decir, para genes con un log2FC < 0 y el rojo para genes con un log2FC > 0.
Conclusión
En este tutorial, hemos analizado datos reales de secuenciación de ARN para extraer información útil, como qué genes están regulados al alza o a la baja por la depleción del gen Pasilla, pero también en qué términos GO o vías KEGG están implicados. Para responder a estas preguntas, analizamos conjuntos de datos de secuencias de ARN utilizando un enfoque de análisis de datos de ARN-Seq basado en referencias. Este enfoque puede resumirse con el siguiente esquema:
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Levin, J. Z., M. Yassour, X. Adiconis, C. Nusbaum, D. A. Thompson et al., 2010 Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods 7: 709. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3005310/
Brooks, A. N., L. Yang, M. O. Duff, K. D. Hansen, J. W. Park et al., 2011 Conservation of an RNA regulatory map between Drosophila and mammals. Genome Research 21: 193–202. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3032923/
Kim, D., B. Langmead, and S. L. Salzberg, 2015 HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12: 357. https://www.nature.com/articles/nmeth.3317
Ewels, P., M. Magnusson, S. Lundin, and M. K\~ A\textcurrencyller, 2016 MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048. 10.1093/bioinformatics/btw354
Kim, D., J. M. Paggi, C. Park, C. Bennett, and S. L. Salzberg, 2019 Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37: 907–915. https://www.nature.com/articles/s41587-019-0201-4
Retroalimentación
¿Utilizaste este material como instructor? Cuéntanos tu experiencia.
¿Has usado este material como aprendiz o estudiante? Haz click en el formulario a continuación para dejarnos tu opinión
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{transcriptomics-ref-based,
author = "Bérénice Batut and Mallory Freeberg and Mo Heydarian and Anika Erxleben and Pavankumar Videm and Clemens Blank and Maria Doyle and Nicola Soranzo and Peter van Heusden and Lucille Delisle",
title = "Análisis de datos RNA-Seq basados en referencias (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/ref-based/tutorial_ES.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Referencias
These individuals or organisations provided funding support for the development of this resource
Preguntas:

Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab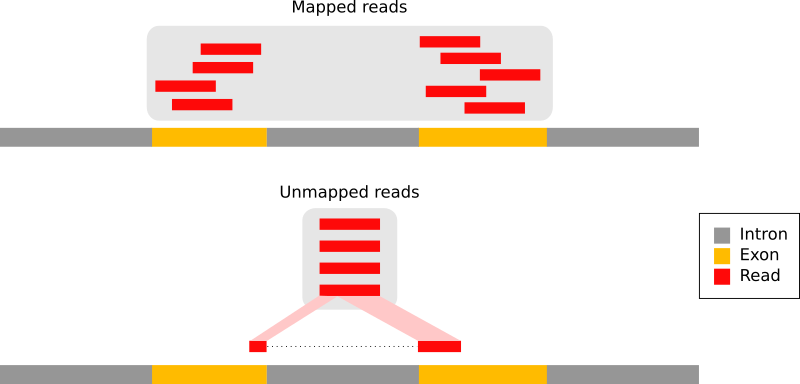 Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab

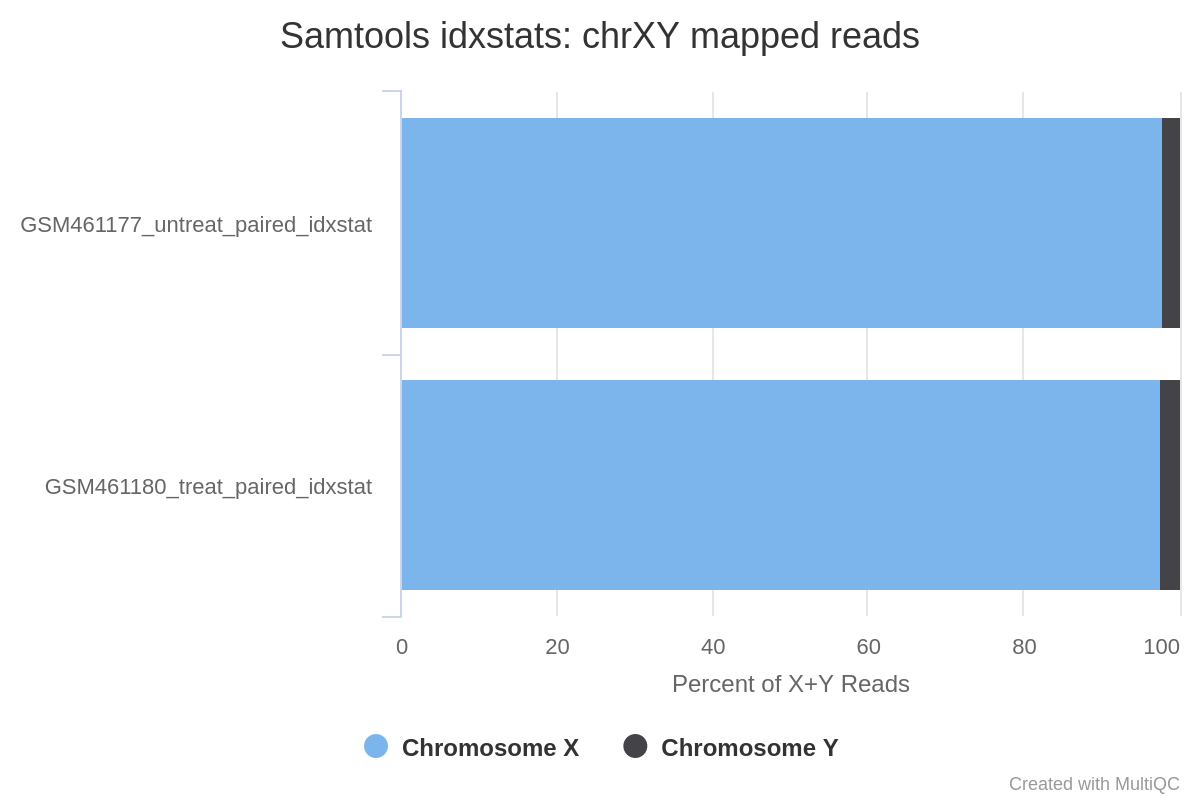
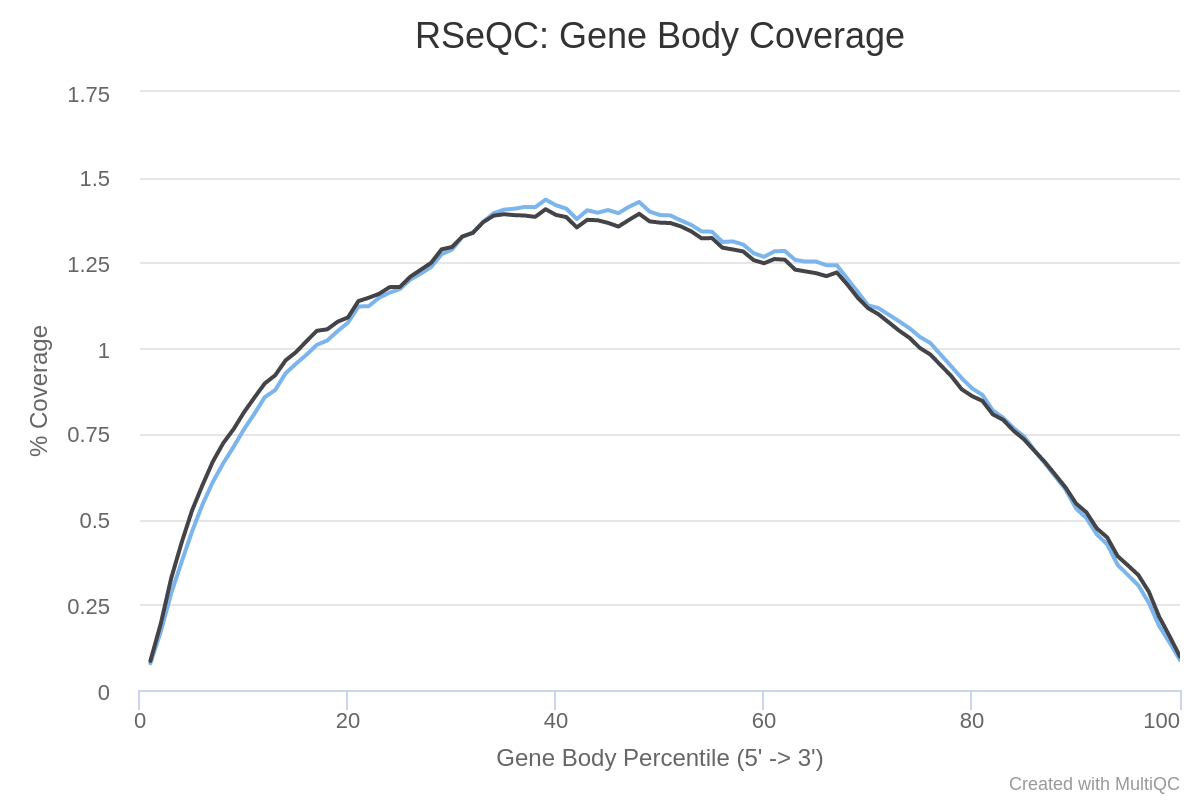
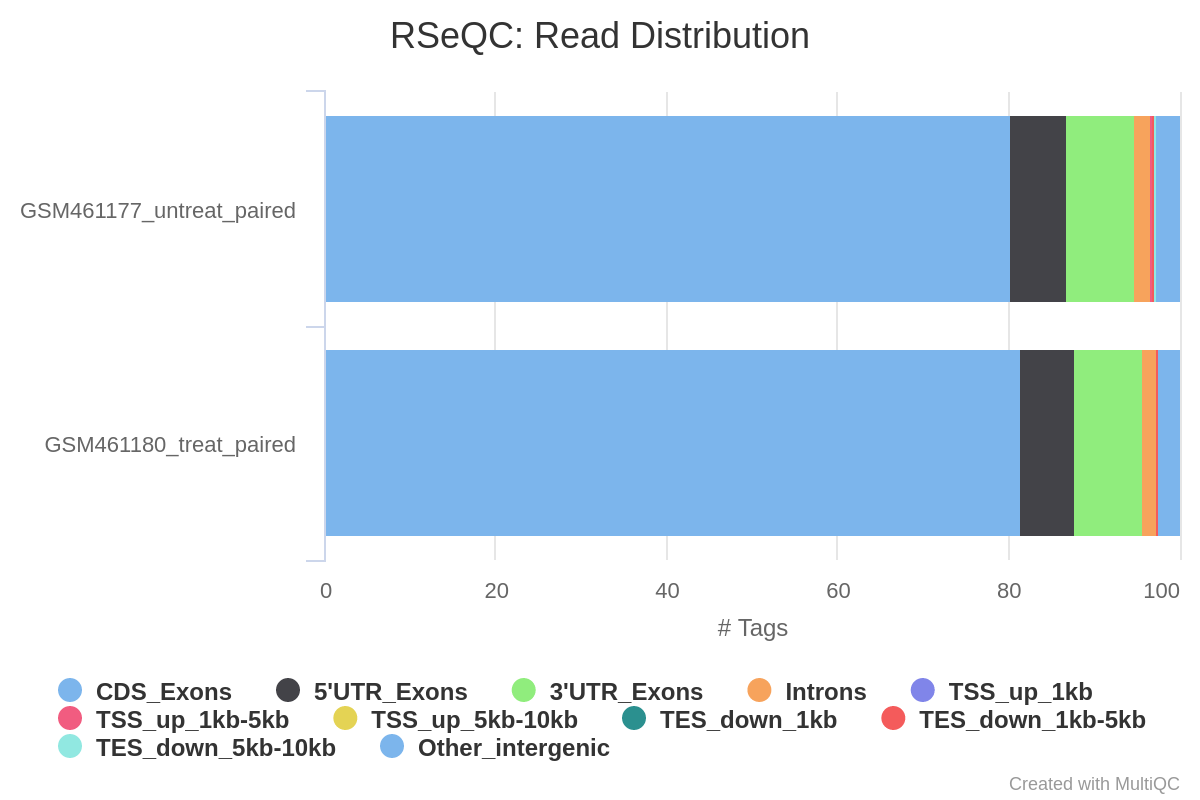
 Open image in new tab
Open image in new tab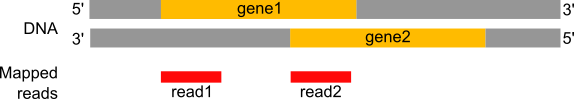 Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
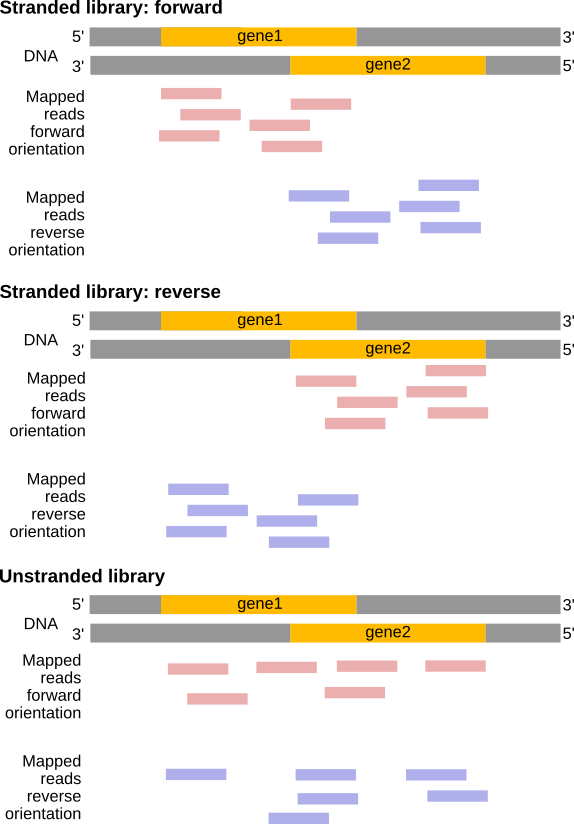 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab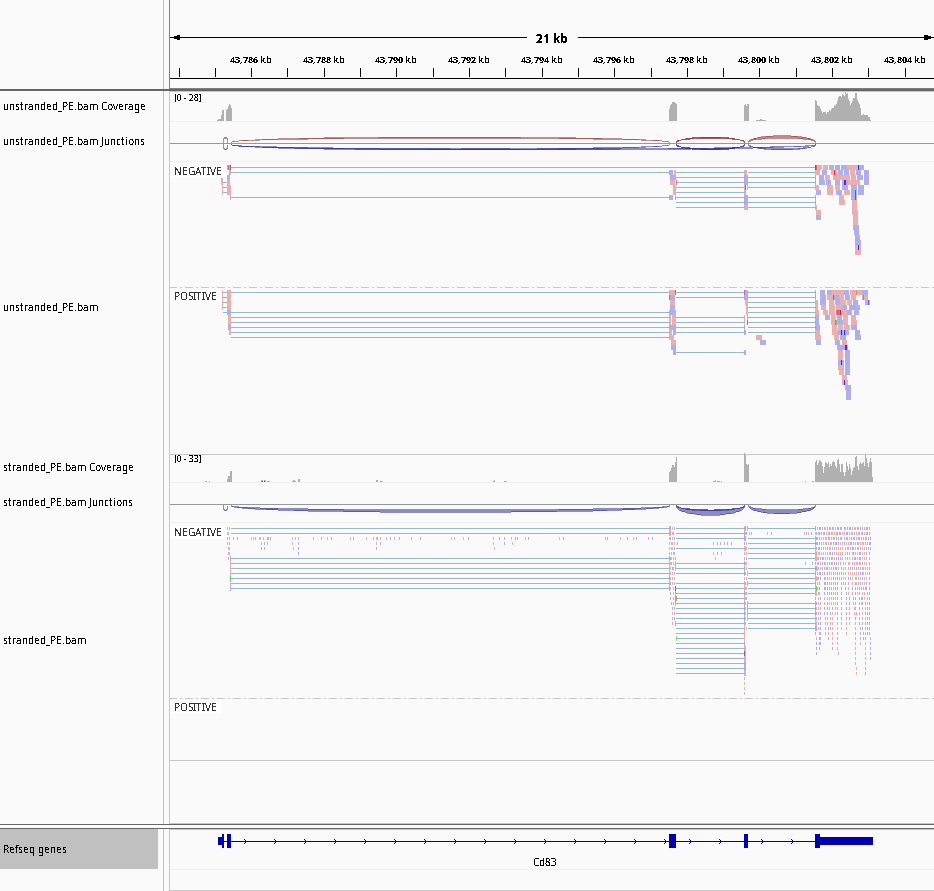 Open image in new tab
Open image in new tab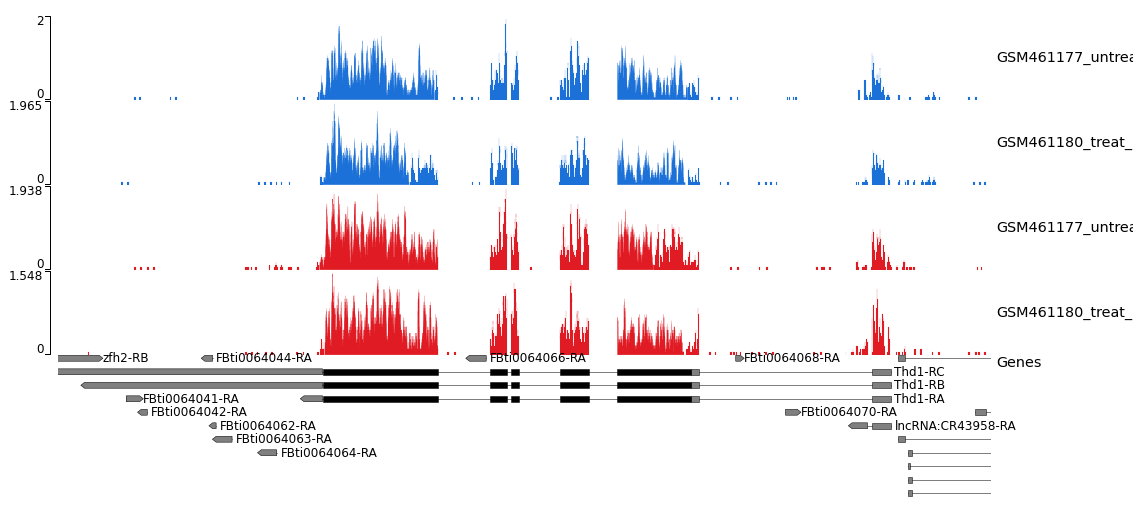 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab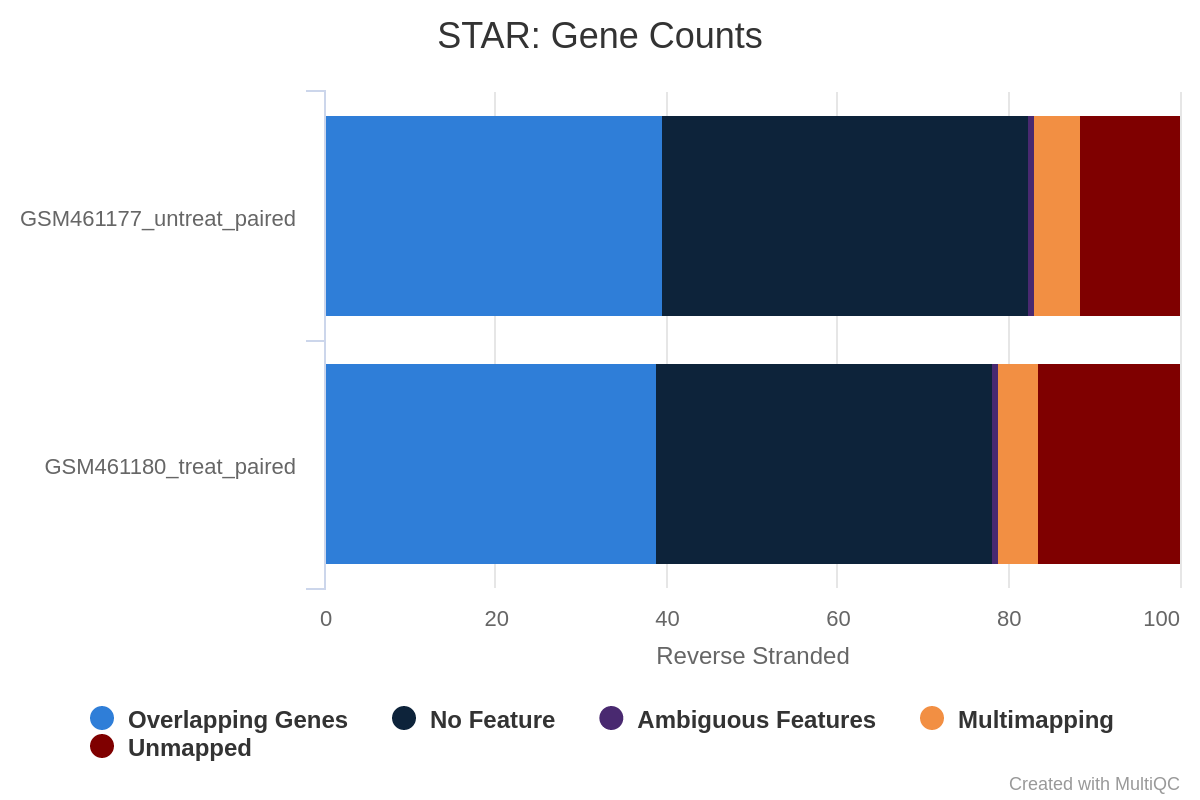 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab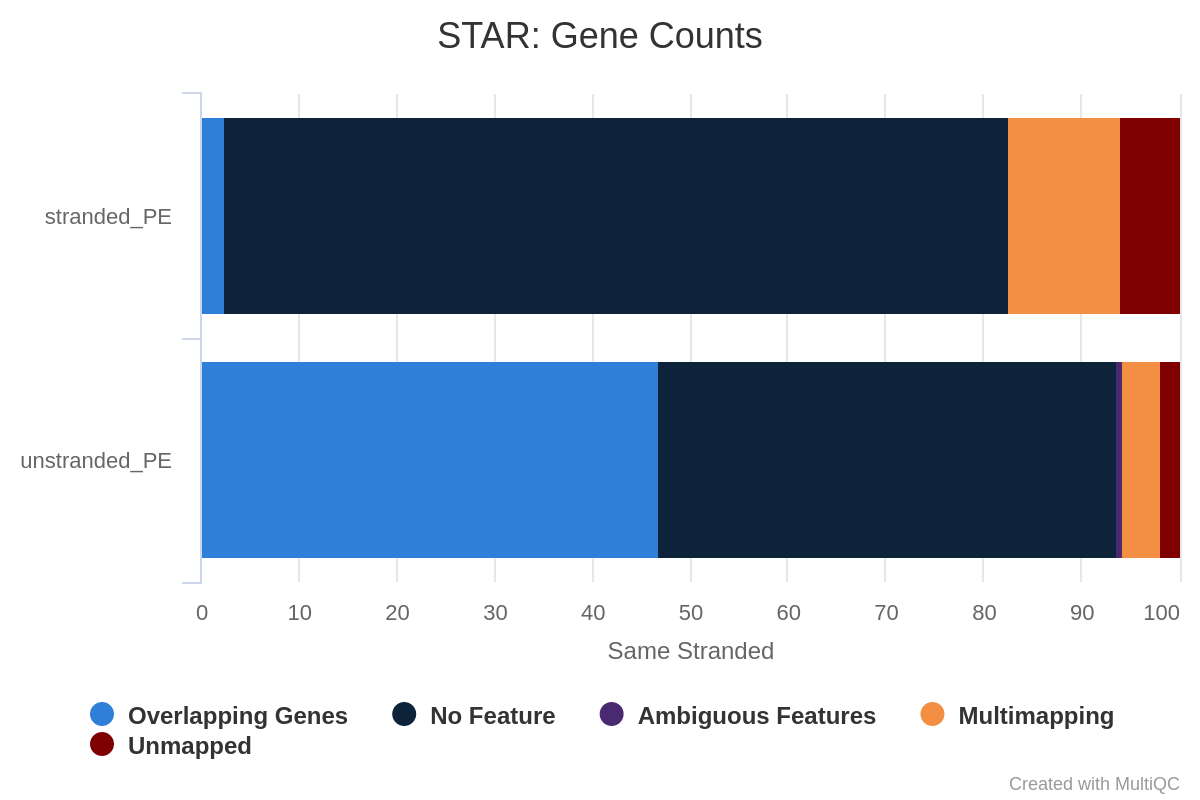 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
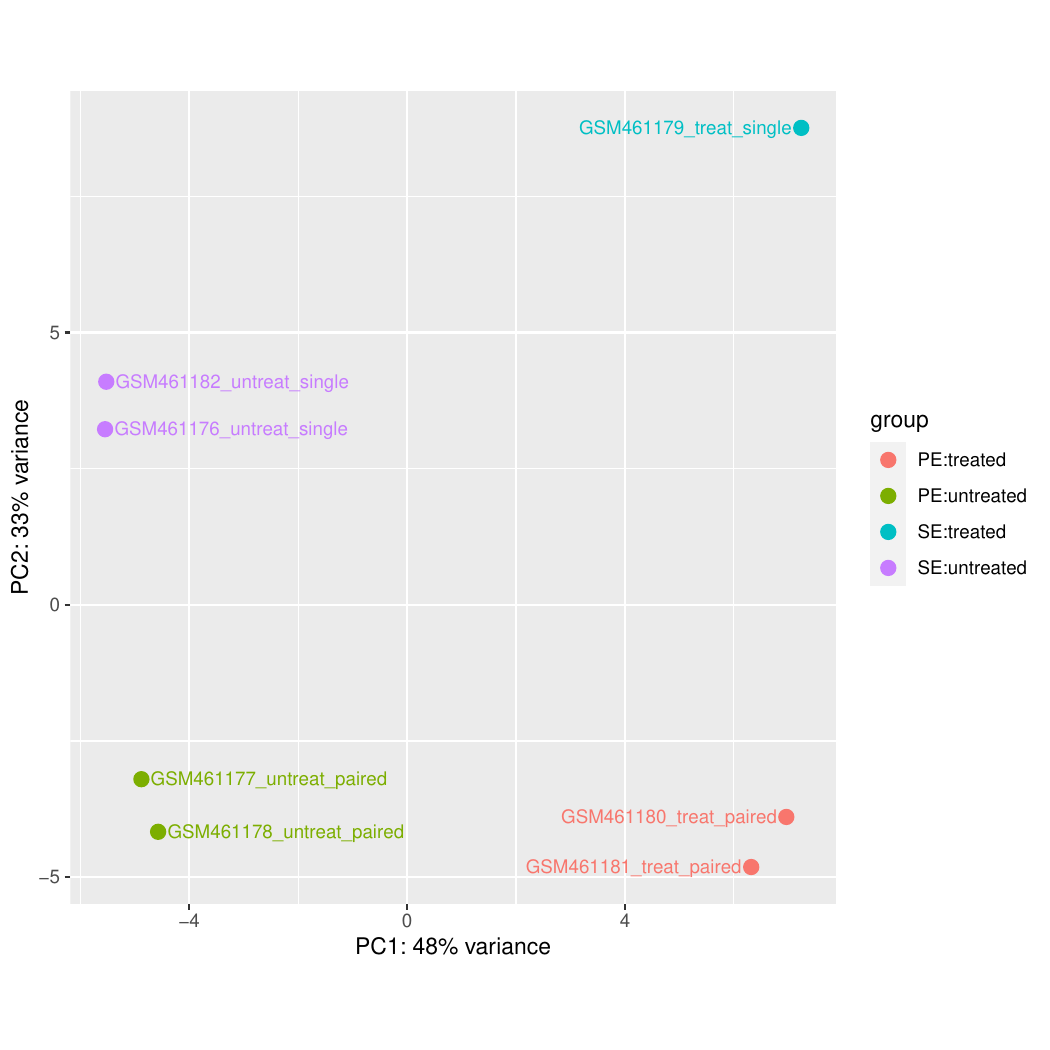 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab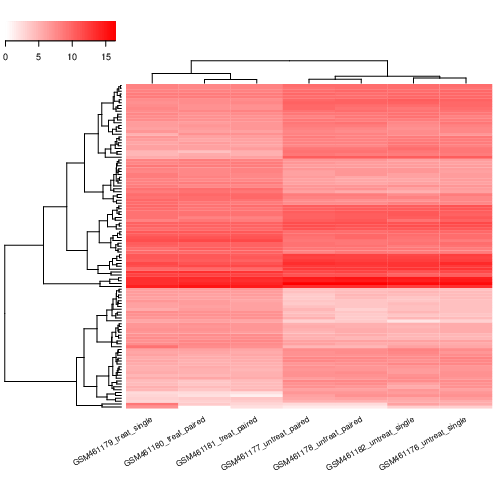 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab


 Open image in new tab
Open image in new tab
