Negli ultimi anni, il sequenziamento dell’RNA (RNA-Seq) è diventata una tecnologia ampiamente utilizzata per analizzare il trascrittoma cellulare in continua evoluzione, ovvero l’insieme di tutte le molecole di RNA presenti in una cellula o in una popolazione di cellule. Uno degli obiettivi più comuni dell’RNA-Seq è la profilazione dell’espressione genica, ossia l’identificazione dei geni o delle vie molecolari che risultano differenzialmente espressi (DE) tra due o più condizioni biologiche. Questo tutorial illustra un flusso di lavoro computazionale per l’individuazione di geni e percorsi DE a partire da dati di RNA-Seq, fornendo un’analisi completa di un esperimento RNA-Seq che profila cellule di Drosophila dopo la deplezione di un gene regolatore.
Nello studio di Brooks et al. 2011, gli autori hanno identificato geni e vie molecolari regolati dal gene Pasilla (l’omologo drosophila delle proteine Nova-1 e Nova-2 regolatori dello splicing nei mammiferi) utilizzando dati di RNA-Seq. Il gene Pasilla (PS) in Drosophila melanogaster è stato silenziato mediante il metodo dell’interferenza a RNA (RNAi). L’RNA totale è stato quindi isolato e utilizzato per preparare librerie RNA-Seq di tipo single-end e paired-end per i campioni trattati (privi di PS) e non trattati. Le librerie sono state successivamente sequenziate per ottenere le letture RNA-Seq per ciascun campione. I dati RNA-Seq dei campioni trattati e non trattati possono essere confrontati per identificare gli effetti della deplezione del gene Pasilla sull’espressione genica.
In questa esercitazione, illustriamo l’analisi dei dati di espressione genica passo dopo passo utilizzando 7 set di dati originali:
Ogni campione rappresenta un replicato biologico indipendete della condizione corrispondente (trattata o non trattata). Inoltre, due dei campioni trattati e due di quelli non trattati derivano da un saggio di sequenziamento paired-end, mentre i restanti campioni provengono da un esperimento di sequenziamento single-end.
Commento: Dati completi
I dati originali sono disponibili nel database NCBI Gene Expression Omnibus (GEO) con il numero di accesso GSE18508. Le letture RNA-Seq grezze sono state estratte dai file del Sequence Read Archive (SRA) e convertite in file FASTQ.
Nella prima parte di questo tutorial utilizzeremo i file di 2 dei 7 campioni per dimostrare come calcolare il conteggio delle letture (una misura dell’espressione genica) a partire dai file FASTQ (controllo di qualità, mappatura, conteggio delle letture). Forniamo i file FASTQ per gli altri 5 campioni nel caso si desideri riprodurre l’intera analisi in un secondo momento.
Nella seconda parte del tutorial, i conteggi delle letture di tutti e 7 i campioni vengono utilizzati per identificare e visualizzare i geni differenzialmente espressi, le famiglie geniche e le vie molecolari associate alla deplezione del gene PS.
Pratica: Caricamento dati
Creare una nuova storia (sessione di lavoro) per questo tutorial di RNA-Seq
Per creare una nuova storia è sufficiente fare clic sull’icona new-history nella parte superiore del pannello della storia:
Importare le coppie di file FASTQ da Zenodo o da una libreria di dati:
GSM461177 (non trattato): GSM461177_1 e GSM461177_2
Fare clic su galaxy-uploadCarica i dati nella parte superiore del pannello degli strumenti
Selezionare galaxy-wf-editIncollare/recuperare i dati
Incollare il/i link nel campo di testo
Premere Avvio
Chiude la finestra
In alternativa al caricamento dei dati da un URL o dal proprio computer, i file possono essere resi disponibili da una libreria di dati condivisi:
Entrare in Librerie (pannello sinistro)
Navigare verso alla cartella corretta indicata dal vostro istruttore. Nella maggior parte dei Galaxies i dati delle esercitazioni vengono forniti in una cartella denominata GTN - Materiale –> Nome argomento -> Nome esercitazione.
selezionare i file desiderati
Fare clic su Aggiungi alla cronologiagalaxy-dropdown vicino alla parte superiore e selezionare as Datasets dal menu a tendina
Nella finestra pop-up, scegliere
“Seleziona cronologia “: la cronologia in cui si desidera importare i dati (o crearne una nuova)
Cliccare su Import
Commento
Si noti che questi sono i file completi dei campioni, di circa 1,5 GB ciascuno, quindi l’importazione potrebbe richiedere alcuni minuti.
Per un’esecuzione più rapida dei passaggi relativi ai file FASTQ, è disponibile un piccolo sottoinsieme di ciascun file (~5 MB) su Zenodo:
Verifica che il tipo di dato sia fastqsanger (ad esempio nonfastq). In caso contrario, modifica il tipo di dato in fastqsanger.
Cliccare sull’icona galaxy-pencilicona della matita per il set di dati per modificarne gli attributi
Nel pannello centrale, fare clic su galaxy-chart-select-data *scheda *Datipi** in alto
Nella sezione galaxy-chart-select-dataAssegna tipo di dato, selezionare fastqsanger dal menu a discesa “Nuovo tipo”
Suggerimento: si può iniziare a digitare il tipo di dato nel campo per filtrare il menu a discesa
Fare clic sul pulsante Salva
Crea una raccolta di coppie denominata 2 PE fastqs, assegnando alle coppie il nome del campione seguito dagli attributi: GSM461177_untreat_paired e GSM461180_treat_paired.
Fare clic su galaxy-selectorSeleziona elementi nella parte superiore del pannello della cronologia
Controllare tutti i dataset della cronologia che si desidera includere
Fare clic su n di N selezionati e scegliere Costruisci elenco di coppie di set di dati
Cambiare il testo di unpaired forward in un selettore comune per le letture forward
Cambiare il testo di unpaired reverse con un selettore comune per le letture inverse
Fare clic su Accoppia questi set di dati per ogni coppia avanti e inversa valida.
Inserire un nome per la collezione
Fare clic su Crea elenco per creare la raccolta
Fare nuovamente clic sull’icona del segno di spunta in cima alla cronologia
Domanda
Come vengono memorizzate le sequenze di DNA?
Quali sono le altre voci del file?
Le sequenze di DNA sono memorizzate in un file FASTQ, nella seconda riga di ogni gruppo di 4 righe.
Questo formato di file è chiamato formato FASTQ. Memorizza informazioni sulla sequenza e sulla qualità. Ogni sequenza è rappresentata da un gruppo di 4 righe: la prima riga è l’id della sequenza, la seconda è la sequenza dei nucleotidi, la terza è una riga di transizione e l’ultima è una sequenza del punteggio di qualità per ciascun nucleotide.
Le letture rappresentano dati grezzi provenienti dalla macchina di sequenziamento senza alcun pretrattamento. È necessario valutarne la qualità.
Controllo qualità
Durante il sequenziamento possono verificarsi errori, come il richiamo errato di nucleotidi, dovuti alle limitazioni tecniche di ciascuna piattaforma di sequenziamento. Tali errori di sequenziamento possono influenzare l’analisi e portare a un’interpretazione non corretta dei dati. Se le letture sono più lunghe dei frammenti sequenziati, possono contenere degli adattatori, la cui rimozione può migliorare il numero di letture mappate.
Il controllo della qualità delle sequenze è quindi un primo passo essenziale nella vostra analisi. Utilizzeremo strumenti simili a quelli descritti nel tutorial “Controllo di qualità”:
Falco, una riscrittura ottimizzata per l’efficienza di FastQC, per generare un rapporto sulla qualità delle sequenze
Cutadapt (Marcel 2011) per migliorare la qualità delle sequenze attraverso il taglio e il filtraggio.
Purtroppo la versione attuale di MultiQC (lo strumento che utilizziamo per combinare i report) non supporta le raccolte di liste di coppie. Dovremo quindi prima trasformare la nostra raccolta di coppie in una semplice lista.
La situazione attuale è mostrata in alto e lo strumento Flatten collection la trasformerà in quella visualizzata in basso: Open image in new tab
<figcaption>Figura 1: Flatten the list of pairs to list</figcaption>
Pratica: Controllo qualità
Flatten collection con i seguenti parametri, per convertire la raccolta di coppie in un elenco semplice:
“Collezione di input “: 2 PE fastqs
Falco ( Galaxy version 1.2.4+galaxy0) con i seguenti parametri:
param-collection“Dati di lettura grezzi dalla cronologia corrente “: Output dello strumento Flatten collectiontool selezionata come raccolta di dati
Fare clic su param-collectionCollezione di set di dati davanti al parametro di input a cui si vuole fornire la collezione.
Selezionare la collezione che si desidera utilizzare dall’elenco
Ispezionare l’output della pagina web di Falcotool per il campione GSM461177_untreat_paired (forward e reverse)
Domanda
Qual è la lunghezza delle lettura?
La lunghezza delle letture di entrambi gli accoppiamenti è di 37 bp.
Poiché è noioso ispezionare tutti questi rapporti individualmente, li combineremo con MultiQC ( Galaxy version 1.11+galaxy1).
MultiQC ( Galaxy version 1.24.1+galaxy0) per aggregare i rapporti Falco con i seguenti parametri:
In “Risultati “:
“Risultati “
“Quale strumento è stato utilizzato per generare i log? “: FastQC
Falco è un sostituto di FastQC e possiamo passare il suo output a MultiQC come se fosse stato generato dallo strumento originale.
In “FastQC output “:
param-repeat“Inserisci output FastQC “
param-collection“Output FastQC “: Falco on collection N: RawData (output di Falcotool)
Ispeziona la pagina web prodotta da MultiQC per ciascun file FASTQ
Domanda
Cosa ne pensi della qualità delle sequenze?
Cosa dobbiamo fare?
Tutto sembra buono per 3 dei file. Il file GSM461177_untreat_paired contiene 10,6 milioni di sequenze appaiate e il file GSM461180_treat_paired 12,3 milioni di sequenze appaiate. Tuttavia in GSM461180_treat_paired_reverse (letture inverse di GSM461180) la qualità diminuisce sensibilmente verso fine delle sequenze.
Tutti i file, eccetto GSM461180_treat_paired_reverse, mostrano un’elevata percentuale di letture duplicate (atteso nei dati RNA-Seq).
La “qualità della sequenza per base” è globalmente buona, con una leggera diminuzione alla fine delle sequenze. Per il campione GSM461180_treat_paired_reverse, la diminuzione è invece piuttosto marcata.
Non sono presenti adattatori noti e le sequenze sovrarappresentate sono quasi assenti.
Se la qualità delle letture è scarsa, è necessario:
Verificare cosa non va e riflettere sulle possibili cause: la bassa qualità potrebbe dipendere dal tipo di sequenziamento o dal materiale sequenziato (ad esempio un’elevata quantità di sequenze sovrarappresentate nei dati di trascrittomica o una percentuale distorta di basi nei dati Hi-C)
Contattare il centro di sequenziamento per ottenere chiarimenti.
Eseguire un trattamento di qualità (prestando attenzione a non perdere troppe informazioni), ad esempio effettuando un leggero trimming o rimuovendo le letture di scarsa qualità
Dovremmo tagliare le letture per eliminare le basi che sono state sequenziate con un’elevata incertezza, cioè basi di bassa qualità, alle estremità delle letture, e rimuovere anche le letture con una qualità complessiva scarsa.
Domanda
Qual è la relazione tra GSM461177_untreat_paired_forward e GSM461177_untreat_paired_reverse?
I dati sono stati sequenziati utilizzando il sequenziamento paired-end.
Il sequenziamento paired-end si basa sull’idea che i frammenti di DNA iniziali (più lunghi della lunghezza effettiva della lettura) vengano sequenziati da entrambi i lati. Questo approccio dà luogo a due letture per frammento, con la prima lettura in orientamento in avanti e la seconda in orientamento inverso-complementare. La distanza tra le due letture è nota. Pertanto, può essere utilizzata come informazione aggiuntiva per migliorare la mappatura delle letture.
Con il sequenziamento paired-end, ogni frammento è più coperto che con il sequenziamento single-end (viene sequenziato solo l’orientamento forward):
Il sequenziamento paired-end genera quindi 2 file:
Un file con le sequenze corrispondenti all’orientamento in avanti di tutti i frammenti
Un file con le sequenze corrispondenti all’orientamento inverso di tutti i frammenti
Qui GSM461177_untreat_paired_forward corrisponde alle letture in avanti e GSM461177_untreat_paired_reverse alle letture inverse.
Pratica: Trimming FASTQ
Cutadapt ( Galaxy version 4.9+galaxy1) con i seguenti parametri per tagliare le sequenze di bassa qualità:
“Single-end or Paired-end reads? “: Paired-end Collection
param-collection“Paired Collection “: 2 PE fastqs
In “Other Read Trimming Options “
“Quality cutoff(s) (R1) “: 20
In “Read Filtering Options “
“Minimum length (R1) “: 20
In “Additional outputs to generate “
Selezionare: Report: Cutadapt's per-adapter statistics. You can use this file with MultiQC.
Domanda
Perché lo strumento di trimming viene eseguito solo una volta su un set di dati paired-end e non due volte, una per ogni set di dati?
Lo strumento può rimuovere le sequenze che diventano troppo corte durante il processo di trimming. Per i file paired-end rimuove intere coppie di sequenze se una (o entrambe) delle due letture diventa più corta del limite di lunghezza impostato. Le letture di una coppia di letture che sono più lunghe di una determinata soglia, ma per le quali la lettura partner è diventata troppo corta, possono essere facoltativamente scritte in file single-end. In questo modo si garantisce che le informazioni di una coppia di letture non vadano perse del tutto se solo una lettura è di buona qualità.
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i rapporti prodotti da Cutadapt e visualizzare i risultati complessivi:
In “Results “:
param-repeat“Results “
“Which tool was used generate logs? “: Cutadapt/Trim Galore!
param-collection“Output of Cutadapt “: Cutadapt on collection N: Report (output of Cutadapttool) selezionato as Dataset collection
Domanda
Quante coppie di letture sono state rimosse perché almeno una delle due era più corta della soglia di lunghezza?
Quante coppie di basi (bp) sono state rimosse dalle letture forward per bassa qualità? E dalle reverse?
147.810 (1,4%) letture erano troppo corte per GSM461177_untreat_paired e 1.101.875 (9%) per GSM461180_treat_paired. Open image in new tab
LL’output di MultiQC riporta solo la percentuale totale di bp tagliati, non il dettaglio per ciascuna lettura. Per ottenere questa informazione occorre tornare ai report individuali. Per GSM461177_untreat_paired, ono stati tagliati 5.072.810 bp dalle letture forward (Read 1) e 8.648.619 bp dalle letture reverse (Read 2) per motivi di qualità. Per GSM461180_treat_paired, 10.224.537 bp dalle forward e 51.746.850 bp dalle reverse. Non sorprende: abbiamo già osservato un calo più marcato della qualità verso la fine delle letture reverse rispetto alle forward, in particolare per GSM461180_treat_paired.
Mappatura
Per interpretare correttamente le letture, dobbiamo innanzitutto determinare da dove provengono le sequenze all’interno del genoma, così da poter stabilire a quali geni appartengono. Quando è disponibile un genoma di riferimento per l’organismo, questo processo è chiamato allineamento o mappatura delle letture al genoma di riferimento. È un po’ come risolvere un puzzle: tuttavia, non tutti i pezzi sono unici.
Commento
Volete saperne di più sui principi della mappatura? Seguite il nostro training.
In questo studio, gli autori hanno utilizzato cellule di Drosophila melanogaster. Pertanto, le sequenze sottoposte a controllo qualità devono essere mappate sul genoma di riferimento di Drosophila melanogaster.
Domanda
Cos’è un genoma di riferimento?
Per ogni organismo modello, possono essere disponibili diversi genomi di riferimento (per esempio, hg19 e hg38 per l’uomo). A cosa corrispondono?
Quale genoma di riferimento dobbiamo usare?
Un genoma di riferimento (o assemblaggio di riferimento) è un insieme di sequenze di acidi nucleici assemblate come esempio rappresentativo del materiale genetico di una specie. Poiché spesso sono assemblati a partire dal sequenziamento di diversi individui, non rappresentano accuratamente l’insieme dei geni di un singolo organismo, ma un mosaico di diverse sequenze di acidi nucleici provenienti da ciascun individuo.
Con la diminuzione del costo del sequenziamento del DNA e l’emergere di nuove tecnologie di sequenziamento del genoma completo, continuano a essere generate più sequenze di genomi. Utilizzando queste nuove sequenze, si costruiscono nuovi allineamenti e si migliorano i genomi di riferimento (meno lacune, correzione di errori di rappresentazione nella sequenza, ecc.) I diversi genomi di riferimento corrispondono alle diverse versioni rilasciate (chiamate “build”).
Il genoma di Drosophila melanogaster è noto e assemblato e può essere utilizzato come genoma di riferimento per questa analisi. Si noti che nuove versioni dei genomi di riferimento vengono pubblicate quando l’assemblaggio migliora; pin questa esercitazione utilizzeremo la release 6 dell’assemblaggio del genoma di riferimento di Drosophila melanogaster(dm6).
Nei trascrittomi eucariotici, la maggior parte delle letture deriva da mRNA maturi privi di introni:
Figura 9: I tipi di letture RNA-seq (adattamento della Figura 1a da Kim et al. 2015): letture che hanno mappato interamente all'interno di un esone (in rosso), letture che si estendono su 2 esoni (in blu), letture che si estendono su più di 2 esoni (in viola)
Pertanto, queste letture non possono essere semplicemente mappate al genoma come avviene per i dati di DNA. Per gestire correttamente i punti di giunzione tra esoni, sono stati sviluppati strumenti di mappatura “splicing-aware” (consapevoli dello splicing), in grado di allineare in modo efficiente le letture derivate dai trascritti rispetto al genoma di riferimento:
Figura 10: Principio dei mappatori dello splicing: (1) identificazione delle letture che coprono un singolo esone, (2) identificazione delle giunzioni di splicing sulle letture non mappate
Negli ultimi anni sono stati sviluppati diversi spliced mappers per affrontare l’enorme quantità di dati RNA-Seq.
TopHat (Trapnell et al. 2009è stato uno dei primi strumenti creati appositamente per questo scopo. In TopHat le letture vengono allineate al genoma e suddivise in due categorie:: (1) quelle che si mappano direttamente e (2) quelle inizialmente non mappate (IUM).Le “pile” di letture che rappresentano potenziali esoni vengono estese alla ricerca di siti di splicing donatore/accettore e le possibili giunzioni vengono ricostruite. Le letture IUM vengono quindi riallineate su queste giunzioni.
Per ottimizzare e velocizzare ulteriormente l’allineamento delle letture spliced, è stato sviluppato HISAT2 (Kim et al. 2019). UUtilizza un indice FM FM a grafo gerarchic (HGFM), che rappresenta l’intero genoma (e le sue varianti), combinato con indici locali sovrapposti (~57 kb ciascuno). Questo approccio permette di individuare rapidamente i punti iniziali di allineamento con l’indice globale e di rifinire gli allineamenti con gli indici locali:
Figura 13: Grafico gerarchico indice FM in HISAT/HISAT2 (Figura S8 da Kim et al. 2015)
Una parte della lettura (freccia blu)iene inizialmente mappata sul genoma utilizzando l’indice FM globale. HISAT2 tenta poi di estendere l’allineamento utilizzando direttamente la sequenza del genoma (freccia viola). Nel caso (a)l’operazione ha successo e la lettura viene allineata completamente all’interno di un esone. Nel caso (b) l’estensione incontra un mismatch. In questa situazione, HISAT2 utilizza l’indice FM locale che si sovrappone a tale posizione per trovare la mappatura appropriata per la parte restante della lettura (freccia verde). Il caso (c) mostra una combinazione di entrambe le strategie: l’inizio della lettura viene mappato utilizzando l’indice FM globale (freccia blu), esteso fino alla fine dell’esone (freccia viola), quindi mappato con l’indice FM locale (freccia verde) e infine ulteriormente esteso (freccia viola).
STAR aligner (Dobin et al. 2013)è un’alternativa veloce per mappare le letture RNA-Seq rispetto a un genoma di riferimento utilizzando un [suffix array] non compresso (https://en.wikipedia.org/wiki/Suffix_array). Opera in due fasi. Nella prima fase esegue una ricerca dei semi
In questo passaggio, una lettura è divisa tra due esoni consecutivi. STAR cerca un prefisso massimo mappabile (MMP) dall’inizio della lettura fino a quando non riesce più a trovare una corrispondenza continua. DSuccessivamente, ricerca un MMP per la parte non allineata della lettura (a). In presenza di mismatch (b) o di regioni non allineabili (c) gli MMP fungono da ancore da cui estendere gli allineamenti.
Nella seconda fase, STAR ricuce gli MMP per generare allineamenti a livello di lettura che, a differenza degli MMP, possono contenere mismatch e indel. Viene utilizzato uno schema di punteggio per valutare e dare priorità alle combinazioni di ricucitura e per gestire le letture che si mappano in più posizioni. STARè estremamente veloce, ma richiede una notevole quantità di memoria RAM per funzionare in modo efficiente.
Mappatura
Mapperemo ora le nostre letture sul genoma di Drosophila melanogaster utilizzando STAR (Dobin et al. 2013).
Pratica: Mappatura degli spliced
Importare l’annotazione genica Ensembl per Drosophila melanogaster (Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz) dalla libreria Shared Data, se disponibile, oppure da Zenodo nella storia di Galaxy corrente.
Verificare che il tipo di dato sia gtf e non gff e che il database sia dm6
Commento: Come ottenere il file di annotazione?
I file di annotazione degli organismi modello possono essere disponibili nella libreria Shared Data (il percorso può variare da un server Galaxy all’altro). È anche possibile scaricarli dall’UCSC, utilizzando lo strumento UCSC Main.
Per generare questo file specifico, il file di annotazione è stato scaricato da Ensembl, che fornisce un database più completo di trascritti, e adattato per essere compatibile con il genoma dm6 installato sui server Galaxy.
RNA STAR ( Galaxy version 2.7.11a+galaxy0) con i seguenti parametri per mappare le letture sul genoma di riferimento:
“Letture single-end o paired-end “: Paired-end (as collection)
param-collection“RNA-Seq FASTQ/FASTA paired reads “: Cutadapt on collection N: Reads (output di Cutadapttool)
“Custom or built-in reference genome “: Use a built-in index
“Reference genome with or without an annotation” “: use genome reference without builtin gene-model but provide a gtf
“Select reference genome “: Fly (Drosophila melanogaster): dm6 Full
param-file“Gene model (gff3,gtf) file for splice junctions”: Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
“Length of the genomic sequence around annotated junctions”: 36
Questo parametro deve corrispondere alla lunghezza delle letture - 1
“Per gene/transcript output”: Per gene read counts (GeneCounts)
“Compute coverage “:
Yes in bedgraph format
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i log di STAR con i seguenti parametri:
In “Results “:
“Results “
“Which tool was used generate logs? “: STAR
In “STAR output “:
param-repeat“Insert STAR output “
“Type of STAR output? “: Log
param-collection“STAR log output “: RNA STAR on collection N: log (output of RNA STARtool)
Domanda
Quale percentuale di letture è mappata esattamente una volta per entrambi i campioni?
Quali altre statistiche sono disponibili?
Più dell’83% per GSM461177_untreat_paired e del 79% per GSM461180_treat_paired. È possibile procedere con l’analisi, poiché solo percentuali inferiori al 70% devono essere esaminate per verificare una potenziale contaminazione.
Sono disponibili anche il numero e la percentuale di letture mappate in più posizioni, di letture mappate in troppe posizioni diverse e di letture non mappate perché troppo corte.
Probabilmente sarebbe stato possibile essere più rigorosi nella soglia minima di lunghezza delle letture per evitare queste letture non mappate a causa della lunghezza.
Secondo il reporto MultiQC, circa l’80% delle letture di entrambi i campioni si mappa esattamente una volta sul genoma di riferimento. È possibile procedere con l’analisi, poiché solo percentuali inferiori al 70% devono essere verificate per una potenziale contaminazione. Entrambi i campioni mostrano una percentuale bassa (inferiore al 10%) di letture mappate in più posizioni del genoma di riferimento. Questo valore rientra nella norma per i dati di sequenziamento Illumina a lettura breve, ma può essere inferiore per i nuovi dataset di sequenziamento a lettura lunga, che possono coprire regioni ripetute più ampie nel genoma di riferimento, e più elevato per le librerie 3’-end.
L’output principale di STAR è un file BAM.
A BAM (Binary Alignment Map) file is a compressed binary file storing the read sequences, whether they have been aligned to a reference sequence (e.g. a chromosome), and if so, the position on the reference sequence at which they have been aligned.
Pratica: Inspect a BAM/SAM file
Inspect the param-file output of RNA STARtool
A BAM file (or a SAM file, the non-compressed version) consists of:
A header section (the lines starting with @) containing metadata particularly the chromosome names and lengths (lines starting with the @SQ symbol)
An alignment section consisting of a table with 11 mandatory fields, as well as a variable number of optional fields:
Col
Field
Type
Brief Description
1
QNAME
String
Query template NAME
2
FLAG
Integer
Bitwise FLAG
3
RNAME
String
References sequence NAME
4
POS
Integer
1- based leftmost mapping POSition
5
MAPQ
Integer
MAPping Quality
6
CIGAR
String
CIGAR String
7
RNEXT
String
Ref. name of the mate/next read
8
PNEXT
Integer
Position of the mate/next read
9
TLEN
Integer
Observed Template LENgth
10
SEQ
String
Segment SEQuence
11
QUAL
String
ASCII of Phred-scaled base QUALity+33
Domanda
Which information do you find in a SAM/BAM file?
What is the additional information compared to a FASTQ file?
Sequences and quality information, like a FASTQ
Mapping information, Location of the read on the chromosome, Mapping quality, etc
Ispezione dei risultati della mappatura
Il file BAM contiene informazioni su tutte le letture, il che lo rende difficile da ispezionare o esplorare in formato di testo. Uno strumento potente per visualizzare il contenuto dei file BAM è l’Integrative Genomics Viewer (IGV, Robinson et al. 2011).
Fare clic sulla raccolta RNA STAR on collection N: mapped.bam (output di RNA STARtool)
Espandere il param-file file GSM461177_untreat_paired.
Fare clic sull’icona galaxy-barchart visualizza nel blocco file GSM461177.
Nel pannello centrale fare clic su local in display with IGV (local, D. melanogaster (dm6)) per caricare le letture nel browser IGV
Commento
Perché questo passaggio funzioni, è necessario avere installato IGV o Java Web Start sul proprio computer. Tuttavia, le domande di questa sezione possono essere risolte anche osservando le schermate di IGV riportate di seguito.
Figura 17: Screenshot of a Sashimi plot of Chromosome 4
Cosa rappresenta il grafico verticale a barre rosse? E gli archi con i numeri?
Cosa significano i numeri sugli archi?
Perché si osservano diversi gruppi impilati di caselle blu collegate in basso?
La copertura per ogni traccia di allineamento è rappresentata da un grafico a barre rosse. Gli archi rappresentano le giunzioni di splice osservate, cioè le letture che coprono gli introni.
I numeri si riferiscono al numero di letture di giunzione osservate.
I diversi gruppi di caselle collegate nella parte inferiore rappresentano i diversi trascritti dei geni presenti in quella posizione nel file GTF.
La qualità dei dati e della mappatura può essere controllata ulteriormente, ad esempio ispezionando il livello di duplicazione delle letture, il numero di letture mappate su ciascun cromosoma, la copertura del corpo genico e la distribuzione delle letture tra le caratteristiche.
Le letture duplicate possono derivare da geni altamente espressi, quindi di solito vengono mantenute nelle analisi di espressione differenziale RNA-Seq. uttavia, una percentuale elevata di duplicati può indicare un problema, ad esempio un’eccessiva amplificazione durante la PCR di una libreria a bassa complessità.
Il tool MarkDuplicates della Picard suite esamina i record allineati di un file BAM per individuare le letture duplicate, ossia quelle che mappano nella stessa posizione (in base alla posizione iniziale della mappatura).
Pratica: Controllo delle letture duplicate
MarkDuplicates ( Galaxy version 2.18.2.4) con i seguenti parametri:
param-collection“Seleziona il set di dati SAM/BAM o la raccolta di dati “: RNA STAR on collection N: mapped.bam (output di RNA STARtool)
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i log di MarkDuplicates con i seguenti parametri:
In “Results “:
“Results “
“Which tool was used generate logs? “: Picard
In “Picard output “:
param-repeat“Insert Picard output “
“Type of Picard output? “: Markdups
param-collection“Picard output “: MarkDuplicates on collection N: MarkDuplicate metrics (output di MarkDuplicatestool)
Domanda
Quali sono le percentuali di letture duplicate per ciascun campione?
Il campione GSM461177_untreat_paired ha il 25,9% di letture duplicate mentre GSM461180_treat_paired ha il 27,8%.
In generale, ottenere fino al 50% di letture duplicate è considerato normale. Quindi entrambi i nostri campioni vanno bene.
Numero di letture mappate su ciascun cromosoma
Per valutare la qualità dei campioni (ad esempio un eccesso di contaminazione mitocondriale), possiamo controllare il sesso dei campioni o verificare se alcuni cromosomi presentano geni altamente espressi. Questo può essere fatto controllando il numero di letture mappate su ciascun cromosoma utilizzando IdxStats della suite Samtools.
Pratica: Controlla il numero di letture mappate su ciascun cromosoma
Samtools idxstats ( Galaxy version 2.0.4) con i seguenti parametri:
param-collection“File BAM “: RNA STAR on collection N: mapped.bam (output di RNA STARtool)
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i log di idxstats con i seguenti parametri:
In “Results “:
“Results “
“Which tool was used generate logs? “: Samtools
In “Samtools output “:
param-repeat“Insert Samtools output “
“Type of Samtools output? “: idxstats
param-collection“Samtools idxstats output “: Samtools idxstats on collection N (output di Samtools idxstatstool)
Domanda
Quanti cromosomi ha il genoma di Drosophila?
Dove sono state mappate principalmente le letture?
Possiamo determinare il sesso dei campioni?
Il genoma di Drosophila ha 4 coppie di cromosomi: X/Y, 2, 3 e 4.
Le letture sono state mappate principalmente sui cromosomi 2 (chr2L e chr2R), 3 (chr3L e chr3R) e X. Solo poche letture sono state mappate sul cromosoma 4, il che è prevedibile dato che questo cromosoma è molto piccolo.
A giudicare dalla percentuale di letture X+Y, la maggior parte delle letture mappa su X e solo poche su Y. Questo indica che probabilmente non ci sono molti geni su Y, quindi i campioni sono probabilmente entrambi di sesso femminile.
Copertura del corpo genico
LLe diverse regioni di un gene costituiscono il corpo del gene. È importante verificare se la copertura delle letture è uniforme lungo tutto il corpo genico. Ad esempio, un bias verso l’estremità 5’ può indicare una degradazione dell’RNA, mentre un bias verso il 3’ può indicare che i dati provengono da un saggio 3’. Per valutare questo aspetto, si utilizza lo strumento Gene Body Coverage della suite di strumenti RSeQC (Wang et al. 2012). uesto strumento ridimensiona tutti i trascritti a 100 nucleotidi (usando un file di annotazione fornito) e calcola il numero di letture che coprono ciascuna posizione (scalata) del trascritto. Poiché questo strumento è piuttosto lento, la copertura viene calcolata solo su 200.000 letture casuali.
Pratica: Controllo della copertura del corpo genico
Samtools view ( Galaxy version 1.15.1+galaxy0) con i seguenti parametri:
param-collection“Set di dati SAM/BAM/CRAM “: mapped_reads (output di RNA STARtool)
“Cosa vorresti guardare? “: A filtered/subsampled selection of reads
In “CConfigure subsampling “:
“ASubsample alignment “: Specify a target # of reads]
“Target # of reads “: 200000
“Seed for random number generator “: 1
“What would you like to have reported? “: All reads retained after filtering and subsampling
“Output format “: BAM (-b)
“Use a reference sequence “: No
Convert GTF to BED12 ( Galaxy version 357) per convertire il file GTF in BED:
param-file“File GTF da convertire “: Drosophila_melanogaster.BDGP6.32.109.gtf.gz
Gene Body Coverage (BAM) ( Galaxy version 5.0.1+galaxy2) con i seguenti parametri:
“Eseguire ogni campione separatamente o combinare più campioni in un unico grafico “: Run each sample separately
param-collection“File .bam di ingresso “: output di Samtools viewtool
param-file“Reference gene model “: Convert GTF to BED12 on data N: BED12 (risultato di Converti GTF in BED12tool)
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i risultati di RSeQC con i seguenti parametri:
In “Results “:
“Results “
“Which tool was used generate logs? “: RSeQC
In “RSeQC output “:
param-repeat“Insert RSeQC output “
“Type of RSeQC output “: gene_body_coverage
param-collection“RSeQC gene_body_coverage output “: Gene Body Coverage (BAM) on collection N (text) (output di Gene Body Coverage (BAM)tool)
Domanda
Come appare la copertura lungo i corpi genici? I campioni sono polarizzati in 3’ o 5’?
Per entrambi i campioni la copertura è abbastanza uniforme dalle estremità 5’ alle 3’, nonostante un po’ di rumore al centro. Quindi non si osservano bias evidenti.
Distribuzione delle letture tra le caratteristiche
Nei dati RNA-Seq, ci si aspetta che la maggior parte delle letture si mappi sugli esoni, piuttosto che su introni o regioni intergeniche. Prima di procedere con il conteggio e con l’analisi dell’espressione differenziale, può essere utile controllare la distribuzione delle letture tra le diverse caratteristiche geniche (esoni, CDS, 5’UTR, 3’UTR, introni, regioni intergeniche). Un’elevata percentuale di letture mappate nelle regioni intergeniche può indicare contaminazione da DNA.
Per farlo, utilizzeremo lo strumento Read Distribution di RSeQC (Wang et al. 2012), che utilizza il file di annotazione per identificare la posizione delle varie caratteristiche geniche.
Pratica: Verifica il numero di letture mappate su ciascun cromosoma
Read Distribution ( Galaxy version 5.0.1+galaxy2) con i seguenti parametri:
param-collection“File .bam/.sam di input “: RNA STAR on collection N: mapped.bam (output di RNA STARtool)
param-file“Modello genico di riferimento “: File BED12 (risultato di Converti GTF in BED12tool)
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i risultati della Read Distribution con i seguenti parametri:
In “Results “:
“Results “
“Which tool was used generate logs? “: RSeQC
In “RSeQC output”:
param-repeat“Insert RSeQC output “
“Type of RSeQC output? “: read_distribution
param-collection“RSeQC read_distribution output “: Read Distribution on collection N (output di Read Distributiontool)
Domanda
Cosa ne pensi della distribuzione delle letture?
La maggior parte delle letture si mappa sugli esoni (>80%), circa il 2% sugli introni e circa il 5% nelle regioni intergeniche, come ci aspettavamo. Questo conferma che i nostri dati sono effettivamente dati RNA-Seq e che la mappatura è riuscita correttamente.
Ora che abbiamo controllato i risultati della mappatura delle letture, possiamo procedere alla fase successiva dell’analisi.
Dopo la mappatura, disponiamo ora delle informazioni sulla posizione delle letture sul genoma di riferimento e sulla qualità della loro mappatura. Il passo successivo dell’analisi RNA-Seq consiste nella quantificazione del numero di letture mappate sulle diverse caratteristiche genomiche (geni, trascritti, esoni, ecc.).
Commento
La quantificazione dipende sia dal genoma di riferimento (file FASTA) sia dalle annotazioni associate (file GTF). È fondamentale utilizzare un file di annotazione corrispondente alla stessa versione del genoma di riferimento utilizzata per la mappatura (in questo caso dm6), poiché le coordinate cromosomiche dei geni variano tra versioni diverse del genoma.
Qui ci concentreremo sui geni, poiché vogliamo identificare quelli espressi in modo differenziale a seguito della deplezione del gene Pasilla.
Conteggio del numero di letture per gene annotato
er confrontare l’espressione di singoli geni tra condizioni diverse (ad esempio con o senza deplezione di PS), un primo passo essenziale è quantificare il numero di letture per gene, o più specificamente, il numero di letture che mappano sugli esoni di ciascun gene.
Figura 19: Conteggio del numero di letture per gene annotato
Domanda
Nell’immagine precedente,
Quante letture sono state trovate per i diversi esoni?
Quante letture sono state trovate per i diversi geni?
Numero di letture per esone
Exon
Number of reads
gene1 - exon1
3
gene1 - exon2
2
gene2 - exon1
3
gene2 - exon2
4
gene2 - exon3
3
Il gene1 ha 4 letture, non 5, a causa dello splicing dell’ultima lettura (gene1 - esone1 + gene1 - esone2). Il gene2 ha 6 letture, 3 delle quali sono spliced.
Per il conteggio delle letture sono disponibili due strumenti principali: HTSeq-count (Anders et al. 2015) o featureCounts (Liao et al. 2013). Inoltre, STAR permette di contare le letture durante la mappatura: i suoi risultati sono identici a quelli di HTSeq-count. Sebbene questo risultato sia sufficiente per la maggior parte delle analisi, featureCounts offre una maggiore personalizzazione del conteggio delle letture (qualità minima della mappatura, conteggio delle letture invece dei frammenti, conteggio dei trascritti invece dei geni, ecc.)
IIn linea di principio, il conteggio delle letture che si sovrappongono a caratteristiche genomiche è un compito piuttosto semplice. Tuttavia, è necessario determinare la direzionalità (strandness) della libreria. Questo è infatti un parametro di featureCounts. Al contrario, STAR calcola i conteggi per tutte e tre le possibili opzioni di strandness, ma è comunque necessario conoscere questa informazione per estrarre i conteggi corretti.
tima della filiazione (strandness)
Gli RNA analizzati negli esperimenti di RNA-Seq sono in genere a singolo filamento (per esempio mRNA) e quindi possiedono una polarità, con estremità 5’ e 3’ funzionalmente distinte. Durante un esperimento di RNA-Seq, queste informazioni vengono spesso perse dopo la sintesi del cDNA, la selezione delle dimensioni e la preparazione della libreria di sequenziamento. Tuttavia, sapere se la libreria conserva o meno la direzionalità è importante per il conteggio corretto, soprattutto per le letture che si trovano in regioni dove due geni si sovrappongono su filamenti opposti.
Figura 20: Se l'informazione sulla filiazione è stata persa durante la preparazione della libreria, la lettura1 sarà assegnata al gene1 situato sul filamento in avanti ma la lettura2 sarà 'ambigua' in quanto può essere assegnata al gene1 (filamento in avanti) o al gene2 (filamento inverso)
Alcuni protocolli di preparazione delle librerie creano librerie RNA-Seq cosiddette stranded che conservano le informazioni sugli strand (Levin et al. 2010 fornisce un’eccellente panoramica). In pratica, con i protocolli Illumina RNA-Seq è improbabile che si incontrino tutte le possibilità descritte in questo articolo. Molto probabilmente si avrà a che fare con:
Dati RNA-Seq non direzionali (unstranded)
Dati RNA-Seq direzionali (stranded) ottenuti mediante kit specifici per l’isolamento dell’RNA
Figura 21: Relazione tra orientamento del DNA e dell'RNA
L’implicazione dell’RNA-Seq a filamento è che si può distinguere se le letture derivano da trascritti con codifica inversa o in avanti. Nell’esempio seguente, i conteggi per il gene Mrpl43 possono essere stimati in modo efficiente solo in una libreria stranded, poiché la maggior parte di essi si sovrappone al gene Peo1 nell’orientamento inverso:
Figura 22: Allineamento di letture RNA-Seq non a filamento (in alto) rispetto a quelle specifiche del filamento inverso (in basso) (usando IGV, le letture di mappatura in avanti sono rosse e quelle di mappatura inversa sono blu)
A seconda dell’approccio e del fatto che si esegua un sequenziamento single-end o paired-end, ci sono diverse possibilità su come interpretare i risultati della mappatura di queste letture sul genoma:
Figura 23: Effetti dei tipi di libreria RNA-Seq (Figura adattata dalla documentazione Sailfish)
Le informazioni sulla strandness dovrebbero essere fornite insieme ai file FASTQ; in caso contrario, è possibile richiederle al centro di sequenziamento o trovarle nel sito da cui i dati sono stati scaricati o nella pubblicazione associata.
Figura 24: In una libreria stranded forward, le letture mappano principalmente sullo stesso filamento dei geni. In una libreria stranded reverse, le letture si trovano per lo più sul filamento opposto. Con una libreria non filiforme, le letture si trovano sui geni su entrambi i filamenti, indipendentemente dall'orientamento del gene (esempio per una libreria a lettura singola).
Esistono quattro modi principali per stimare la strandness dai risultati di STAR (scegliere quello che si preferisce)
Possiamo fare un’ispezione visiva dei filamenti delle letture su IGV (per i set di dati Paired-end è meno facile che con le letture singole e quando si hanno molti campioni può essere doloroso).
Pratica: Stimare la filiazione con IGV per una libreria paired-end
Aprire il file BAM del campione GSM461177_untreat_paired.
Nessun problema, basta rifare i passaggi precedenti:
Avviare IGV localmente
Fare clic sulla raccolta RNA STAR on collection N: mapped.bam (output di RNA STARtool)
Espandere il param-file file GSM461177_untreat_paired.
Fare clic su local in display with IGV local D. melanogaster (dm6) per caricare le letture nel browser IGV
IGVtool
Zoom su chr3R:9,445,000-9,448,000 (cromosoma 3 tra 9,445 kb e 9,448 kb), sulla traccia mapped.bam
Fare clic con il pulsante destro del mouse e selezionare Color Aligments by -> first-in-pair strand
Fare clic con il pulsante destro del mouse e selezionare Squished
Figura 26: Schermata dell'IGV per non-stranded (in alto) vs. reverse strand-specific (in basso)
Si noti che non ci sono letture nel gruppo POSITIVO per il filamento inverso specifico.
In alternativa, invece di usare la BAM si può usare la copertura dei filamenti generata da STAR. Utilizzando pyGenomeTracks si potrà visualizzare la copertura su ciascun filamento per ogni campione. Questo strumento dispone di numerosi parametri per personalizzare i grafici.
Pratica: Stimare la strandness con pyGenometracks dalla copertura STAR
pyGenomeTracks ( Galaxy version 3.8+galaxy2):
“Region of the genome to plot “: chr4:540,000-560,000
In “Include tracks in your plot “:
param-repeat“Insert Include tracks in your plot “
“Choose style of the track “: Bedgraph track
“Plot title “: È necessario lasciare questo campo vuoto in modo che il titolo del grafico sia il nome del campione.
param-collection“Track file(s) bedgraph format “: Selezionare RNA STAR on collection N: Coverage Uniquely mapped strand 1.
“Color of track “: Selezionare un colore a scelta, ad esempio blu
“Minimum value “: 0
“height “: 3
“Show visualization of data range “: Yes
param-repeat“Insert Include tracks in your plot “
“Choose style of the track “: Bedgraph track
“Plot title “: È necessario lasciare questo campo vuoto in modo che il titolo del grafico sia il nome del campione.
param-collection“Track file(s) bedgraph format “: Selezionare RNA STAR on collection N: Coverage Uniquely mapped strand 2.
“Color of track “: Selezionare un colore a scelta diverso dal primo, ad esempio rosso
“Minimum valu “: 0
“height “: 3
“Show visualization of data range “: Yes
param-repeat“Insert Include tracks in your plot “
“Choose style of the track”: Gene track / Bed track
“Plot title “: Genes
param-file“Track file(s) bed or gtf format “: Selezionare Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
Figura 28: STAR coverage for strand 1 in blue and strand 2 in red for unstranded and reverse stranded library
Si noti che la copertura sullo strand 1 è molto bassa per il campione stranded_PE mentre il gene è forward. Ciò significa che la libreria di stranded_PE è a filamento inverso. Al contrario, per unstranded_PE la scala è paragonabile per entrambi gli strand.
È possibile utilizzare l’output di STAR con i conteggi. Infatti, come spiegato in precedenza, STAR valuta il numero di letture sui geni per i tre possibili scenari: libreria non filiforme, filiforme in avanti o filiforme inverso. La condizione che attribuisce più letture al gene deve essere quella che corrisponde alla vostra libreria.
Pratica: Stimare la strandness con featureCounts
MultiQC ( Galaxy version 1.11+galaxy1) per aggregare i conteggi STAR con i seguenti parametri:
In “Results “:
“Results “
“Which tool was used generate logs? “: STAR
In “STAR output”:
param-repeat“Insert STAR output “
“ype of STAR output? “: Gene counts
param-collection“STAR gene count output “: RNA STAR on collection N: reads per gene (output di RNA STARtool)
Domanda
Qual è la percentuale di letture assegnata ai geni se la libreria è non direzionale / direzionale (stesso filamento) / direzionale (filamento inverso)?
Figura 30: Gene counts unstranded for unstranded and reverse stranded libraryOpen image in new tab
Figura 31: Gene counts same stranded for unstranded and reverse stranded libraryOpen image in new tab
Figura 32: Gene counts reverse stranded for unstranded and reverse stranded library
Si noti che le letture attribuite ai geni per lo stesso filamento sono molto poche. I numeri sono paragonabili tra unstranded e reverse stranded perché pochi geni si sovrappongono su filamenti opposti, ma comunque si passa dal 63,6% (unstranded) al 65% (reverse stranded).
Un’altra opzione è stimare questi parametri con uno strumento chiamato Infer Experiment negli strumenti di RSeQC (Wang et al. 2012).
Questo strumento prende i file BAM dalla mappatura, seleziona un sottocampione di letture e confronta le loro coordinate genomiche e gli strand con quelli del modello genico di riferimento (da un file di annotazione). In base all’orientamento dei geni, è possibile verificare se il sequenziamento è strand-specifico e, in tal caso, stabilire se le letture sono orientate sul filamento forward o reverse.
Pratica: Determinare la direzionalità della libreria usando Infer Experiment
Convert GTF to BED12 ( Galaxy version 357) per convertire il file GTF in BED:
param-file“GTF File to convert “: Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
È possibile che abbiate già convertito questo file BED12 dal set di dati Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz se avete fatto la parte dettagliata sui controlli di qualità. In questo caso, non è necessario ripetere l’operazione una seconda volta
Infer Experiment ( Galaxy version 5.0.3+galaxy0) per determinare la direzionalità della libreria con i seguenti parametri:
param-collection“Input .bam file “: RNA STAR on collection N: mapped.bam (output di RNA STARtool)
param-file“Reference gene model “: File BED12 (risultato di Converti GTF in BED12tool)
“Number of reads sampled “: 200000
Infer Experiment ( Galaxy version 5.0.3+galaxy0) il tool genera un file con informazioni su:
Libreria Paired-end o single-end
Frazione di letture di cui non è riuscita la determinazione
2 righe
Per le letture single-end
Fraction of reads explained by "++,--": frazione di letture assegnate al filamento forward
Fraction of reads explained by "+-,-+": la frazione di letture assegnate al filamento reverse
Per le letture paired-end
Fraction of reads explained by "1++,1--,2+-,2-+": la frazione di letture assegnate al filamento di forward
Fraction of reads explained by "1+-,1-+,2++,2--": la frazione di letture assegnate al filamento reverse
Se le due “Fraction of reads explained by” hanno valori simili, si può concludere che la libreria non è specifica per il filamento (ovvero è non direzionale / unstranded).
Domanda
Quali sono i risultati della “Fraction of the reads explained by” per GSM461177_untreat_paired?
Pensi che il tipo di libreria dei 2 campioni sia stranded o unstranded?
Risultati per GSM461177_untreat_paired:
Commento: I risultati possono variare
I risultati potrebbero essere leggermente diversi da quelli presentati in questo tutorial a causa di versioni diverse di strumenti, dati di riferimento, database esterni o a causa di processi stocastici negli algoritmi.
This is PairEnd Data
Fraction of reads failed to determine: 0.1013
Fraction of reads explained by "1++,1--,2+-,2-+": 0.4626
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4360
quindi il 46,26% delle letture è assegnato al filamento forward e il 43,60% al filamento reverse.
Si trovano statistiche simili per GSM461180_treat_paired, quindi la libreria sembra essere di tipo unstranded per entrambi i campioni.
Commento: Come sarebbe se la libreria fosse direzionale?
Sempre prendendo come esempio i 2 file BAM, otteniamo per le basi non direzionali:
This is PairEnd Data
Fraction of reads failed to determine: 0.0382
Fraction of reads explained by "1++,1--,2+-,2-+": 0.4847
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4771
E per il filamento inverso:
This is PairEnd Data
Fraction of reads failed to determine: 0.0504
Fraction of reads explained by "1++,1--,2+-,2-+": 0.0061
Fraction of reads explained by "1+-,1-+,2++,2--": 0.9435
Poiché a volte è piuttosto difficile scoprire quali impostazioni corrispondono a quelle di altri programmi, la seguente tabella può essere utile per identificare il tipo di libreria:
Library type
Infer Experiment
TopHat
HISAT2
HTSeq-count
featureCounts
Paired-End (PE) - SF
1++,1–,2+-,2-+
FR Second Strand
Second Strand F/FR
yes
Forward (1)
PE - SR
1+-,1-+,2++,2–
FR First Strand
First Strand R/RF
reverse
Reverse (2)
Single-End (SE) - SF
++,–
FR Second Strand
Second Strand F/FR
yes
Forward (1)
SE - SR
+-,-+
FR First Strand
First Strand R/RF
reverse
Reverse (2)
PE, SE - U
undecided
FR Unstranded
default
no
Unstranded (0)
Conteggio delle letture per geni
Hands-on: Choose Your Own Tutorial
Questa è una sezione 'Choose Your Own Tutorial' (CYOT) (nota anche come 'Choose Your Own Analysis' (CYOA)), in cui è possibile scegliere tra più percorsi. Fare clic su uno dei pulsanti sottostanti per selezionare il modo in cui si desidera seguire l'esercitazione
Per contare il numero di letture per gene, offriamo un tutorial parallelo per i due metodi (STAR e featureCounts) che danno risultati molto simili. Quale metodo preferisci?
Poiché in questo tutorial si è scelto di utilizzare la funzione featureCounts, ora eseguiremo featureCounts per contare il numero di letture assegnate a ciascun gene annotato.
Pratica: Conto del numero di letture per gene annotato
featureCounts ( Galaxy version 2.0.3+galaxy2) con i seguenti parametri per contare il numero di letture per gene:
param-collection“Alignment file “: RNA STAR on collection N: mapped.bam (output di RNA STARtool)
“Specify strand information”: Unstranded
“Gene annotation file”: A GFF/GTF file in your history
Alcune letture non sono state assegnate perché erano multimappate; altre sono state assegnate a nessuna caratteristica o a caratteristiche ambigue.
Se la percentuale è inferiore al 50%, si dovrebbe indagare su dove le letture vengono mappate (all’interno dei geni o meno, con IGV) e controllare che l’annotazione corrisponda alla versione corretta del genoma di riferimento.
L’output principale di featureCounts è una tabella con i conteggi, cioè il numero di letture (o frammenti nel caso di letture paired-end) mappate a ciascun gene (in righe, con il loro ID nella prima colonna) nell’annotazione fornita. FeatureCount genera anche i set di dati di output della *feature length. Questo file ci servirà in seguito per eseguire lo strumento goseq.
Dato che si è scelto di utilizzare la versione STAR del tutorial, useremo STAR per contare le letture.
Come descritto in precedenza, durante la mappatura STAR ha contato le letture per ciascun gene riportato nel file di annotazione genica (questa operazione è stata eseguita tramite l’opzione Per gene read counts (GeneCounts)). Tuttavia, l’output prodotto include alcune statistiche iniziali, seguite dai conteggi per ciascun gene, che variano in base al tipo di libreria: la colonna 2 corrisponde a unstranded, la colonna 3 a stranded forward e la colonna 4 a stranded reverse.
Pratica: Ispetta l'output di STAR
Ispezionare i conteggi di GSM461177_untreat_paired nella raccolta RNA STAR on collection N: reads per gene
Domanda
Quante letture non sono mappate o sono multi-mappate?
A quale riga inizia il conteggio dei geni?
Quali sono le diverse colonne?
Quali sono le colonne più interessanti per il nostro set di dati?
Ci sono 1.190.029 letture non mappate e 571.324 letture multimappate.
Inizia alla riga 5 con il gene FBgn0250732.
Ci sono 4 colonne:
ID del gene
Conteggi per RNA-Seq unstranded)
Conteggi per il primo filamento di lettura allineato all’RNA (stranded forward)
Conteggi per il secondo filamento di lettura allineato all’RNA (stranded reverse)
Poiché i nostri dati non sono stranded, utilizzeremo la colonna Gene ID e la seconda colonna del file.
Riformuleremo l’output di STAR in modo che sia simile a quello di featureCounts (o di altri software di conteggio) che prevede solo due colonne, una con gli ID e l’altra con i conteggi.
Pratica: Riformattazione dell'output STAR
Select last ( Galaxy version 1.1.0) righe da un dataset (coda) per rimuovere le prime 4 righe con i seguenti parametri:
param-collection“Text file”: RNA STAR on collection N: reads per gene (output di RNA STARtool)
“Operation “: Keep everything from this line on
“Number of lines”: 5
Cut colonne da una tabella con i seguenti parametri:
“Cut columns”: c1,c2
“Delimited by”: Tab
param-collection“From “: Select last on collection N (risultato di Select lasttool)
Rinominare la raccolta FeatureCount-like files
Più avanti nell’esercitazione avremo bisogno di conoscere la lunghezza di ciascun gene. Questo valore è già incluso tra gli output di FeatureCounts, ma può anche essere ottenuto direttamente dal file di annotazione genica. Poiché il calcolo richiede un po’ di tempo, è consigliabile avviarlo già ora.
Pratica: Determinare la lunghezza dei geni
Gene length and GC content ( Galaxy version 0.1.2) con i seguenti parametri:
“Select a built-in GTF file or one from your history”: Use a GTF from history
param-file“Select a GTF file”: Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
“Analysis to perform”: gene lengths only
Avviso: Controllare la versione dello strumento
Questo funziona solo con la versione 0.1.2 o superiore
Gli strumenti vengono aggiornati frequentemente con nuove versioni. Nella vostra Galassia potrebbero essere disponibili più versioni dello stesso strumento. Per impostazione predefinita, viene visualizzata la versione più recente dello strumento.
Passaggio a una versione diversa di uno strumento:
aprire lo strumento
Fare clic sul logo tool-versions versioni in alto a destra
Selezionare la versione desiderata dall’elenco a discesa
Domanda
Quale feature (caratteristica genica) presenta il numero più alto di conteggi in entrambi i campioni?
Per visualizzare la feature (caratteristica) più abbondantemente rappresentata, è necessario ordinare la tabella dei conteggi. Questo può essere fatto seguendo la procedura riportata di seguito:
Sort ( Galaxy version 1.1.1) con i seguenti parametri:
param-collection“Sort Query “: featureCounts on collection N: Counts (output di featureCountstool)Utilizzare la collezione FeatureCount-like files
“Number of header”: 10
In “1: Column selections”:
“on column”: Column: 2
Questa colonna contiene il numero di letture = conteggi
“in “: Descending order
Ispezione del risultato
Il risultato dell’ordinamento della tabella sulla colonna 2 rivela che FBgn0284245 è la feature con il maggior numero di conteggi (circa 128.740 in GSM461177_untreat_paired e 127.400 in GSM461180_treat_paired).
Il confronto di diversi file di output è più facile se possiamo visualizzare più di un set di dati contemporaneamente. La funzione Scratchbook ci permette di creare una raccolta di set di dati che verranno visualizzati insieme sullo schermo.
Pratica: (Opzionale) Visualizzare i conteggi ordinati usando Scratchbook
Il Scratchbook si attiva facendo clic sull’icona dei nove blocchi che si trova a destra della barra dei menu superiore di Galaxy:
Figura 35: Barra del menu con icona di Scratchbook abilitata
Fare clic sull’icona galaxy-eye (occhio) per visualizzare uno dei file dei sorted counts. Invece di occupare l’intera barra centrale, la vista del set di dati viene ora mostrata in sovrimpressione:
Figura 36: Scratchbook showing one dataset overlay
Fare quindi clic sull’icona galaxy-eye (occhio) sul second sorted counts. Il secondo set di dati va a sovrapporsi al primo, ma è possibile spostare la finestra per vedere i due set di dati uno accanto all’altro:
Per uscire dalla modalità di selezione di Scratchbook, fare nuovamente clic sull’icona Scratchbook. Si può decidere di chiudere le finestre o di ridurle per poterle visualizzare in seguito.
Qui abbiamo contato le letture mappate sui geni per due campioni. È molto interessante ripetere la stessa procedura sugli altri set di dati, soprattutto per verificare come i parametri differiscano in base al diverso tipo di dati (single-end contro paired-end).
Pratica: (Opzionale) Eseguire nuovamente il test sugli altri set di dati
È possibile eseguire lo stesso processo sugli altri file di sequenza disponibili su Zenodo e sulla libreria di dati.
Dati paired-end
GSM461178_1 e GSM461178_2 che possono essere etichettati come GSM461178_untreat_paired
GSM461181_1 e GSM461181_2 che possono essere etichettati come GSM461181_treat_paired
Dati single-end
GSM461176 che può essere etichettato come GSM461176_untreat_single
GSM461179 che può essere etichettato come GSM461179_treat_single
GSM461182 che può essere etichettato come GSM461182_untreat_single
I collegamenti a questi file sono riportati di seguito:
Per i dati single-end, non è necessario appiattire la raccolta prima di eseguire il passaggio con Falco. II parametri di tutti gli strumenti restano invariati, ad eccezione di STAR, per il quale è possibile impostare Length of the genomic sequence around annotated junctions a 74 poiché un set di dati ha letture di 75bp (gli altri sono 44bp e 45bp). Inoltre, è necessario modificare i parametri di FeatureCounts nel caso in cui i dati non siano più accoppiati.
Analisi dell’espressione genica differenziale
Identificazione dei geni differenzialmente espressi
Per identificare l’espressione genica differenziale indotta dalla deplezione di PS, tutti i set di dati, 3 trattati e 4 non trattati,devono essere analizzati seguendo la stessa procedura. Per risparmiare tempo, abbiamo già eseguito per voi i passaggi preliminari.
Il risultato consiste in 7 file di conteggi, ciascuno contenente il numero di letture associate a ogni gene di Drosophila per ciascun campione.
Pratica: Importa tutti i file di conteggio
Creare una new empty history
Per creare una nuova storia è sufficiente fare clic sull’icona new-history nella parte superiore del pannello della storia:
Importare i sette file di conteggio da Zenodo o dalla libreria Shared Data:
Si potrebbe pensare di poter confrontare direttamente i valori di conteggio nei file e calcolare l’entità dell’espressione genica differenziale. Tuttavia, non è così semplice.
Immaginiamo di avere i conteggi RNA-Seq di 3 campioni per un genoma con 4 geni:
Gene
Sample 1 Counts
Sample 2 Counts
Sample 3 Counts
A (2kb)
10
12
30
B (4kb)
20
25
60
C (1kb)
5
8
15
D (10kb)
0
0
1
Il campione 3 presenta un numero di letture superiore rispetto agli altri replicati, indipendentemente dal gene, poiché è stato sequenziato con una maggiore profondità di lettura. Inoltre, il gene B è lungo il doppio del gene A, e ciò può spiegare perché mostra un numero di letture circa doppio, a prescindere dalle repliche.
Il numero di letture sequenziate mappate su un gene dipende quindi da:
la profondità di sequenziamento dei campioni
I campioni sequenziati con maggiore profondità avranno più letture mappate su ciascun gene
la lunghezza del gene
I geni più lunghi avranno più letture che li mappano
Per confrontare i campioni o le espressioni geniche, i conteggi dei geni devono essere normalizzati. Si può usare il TPM (Transcripts Per Kilobase Million).
Queste tre metriche sono utilizzate per normalizzare le tabelle di conteggio per:
profondità di sequenziamento (la parte “Milione”)
lunghezza del gene (la parte “Kilobase”)
Utilizziamo l’esempio precedente per spiegare RPKM, FPKM e TPM.
Per RPKM (Reads Per Kilobase Million),
Calcolo del fattore di scala “per milione”: sommare le letture totali in un campione e dividere questo numero per 1.000.000.
Gene
Sample 1 Counts
Sample 2 Counts
Sample 3 Counts
A (2kb)
10
12
30
B (4kb)
20
25
60
C (1kb)
5
8
15
D (10kb)
0
0
1
Total reads
35
45
106
Scaling factor
3.5
4.5
10.6
A causa dei piccoli valori nell’esempio, stiamo usando “per decine” invece che “per milioni” e quindi dividiamo la somma per 10 invece che per 1.000.000.
Dividere il conteggio delle letture per il fattore di scala “per milione”
Normalizza per la profondità di sequenziamento, fornendo letture per milione (RPM)
Gene
Sample 1 RPM
Sample 2 RPM
Sample 3 RPM
A (2kb)
2.86
2.67
2.83
B (4kb)
5.71
5.56
5.66
C (1kb)
1.43
1.78
1.43
D (10kb)
0
0
0.09
Nell’esempio usiamo il fattore di scala “per decine” e otteniamo le letture per decine
Dividere i valori RPM per la lunghezza del gene, in kilobasi.
Gene
Sample 1 RPKM
Sample 2 RPKM
Sample 3 RPKM
A (2kb)
1.43
1.33
1.42
B (4kb)
1.43
1.39
1.42
C (1kb)
1.43
1.78
1.42
D (10kb)
0
0
0.009
FPKM (Fragments Per Kilobase Million) è molto simile a RPKM. L’RPKM si usa per l’RNA-seq single-end, mentre l’FPKM si usa per l’RNA-seq paired-end. Con il single-end, ogni lettura corrisponde a un singolo frammento che è stato sequenziato. Con l’RNA-seq paired-end, due letture di una coppia sono mappate da un singolo frammento, oppure se una lettura della coppia non è stata mappata, una lettura può corrispondere a un singolo frammento (nel caso in cui si decida di mantenerle). FPKM tiene traccia dei frammenti in modo che un frammento con 2 letture venga contato una sola volta.
TPM (Transcripts Per Kilobase Million) è molto simile a RPKM e FPKM, tranne che per l’ordine delle operazioni
Dividere il conteggio delle letture per la lunghezza di ciascun gene in kilobasi
Questo dà le letture per kilobase (RPK).
Gene
Sample 1 RPK
Sample 2 RPK
Sample 3 RPK
A (2kb)
5
6
15
B (4kb)
5
6.25
15
C (1kb)
5
8
15
D (10kb)
0
0
0.1
Calcolo del fattore di scala “per milione”: sommare tutti i valori RPK in un campione e dividere questo numero per 1.000.000
Gene
Sample 1 RPK
Sample 2 RPK
Sample 3 RPK
A (2kb)
5
6
15
B (4kb)
5
6.25
15
C (1kb)
5
8
15
D (10kb)
0
0
0.1
Total RPK
15
20.25
45.1
Scaling factor
1.5
2.03
4.51
Come sopra, a causa dei piccoli valori nell’esempio, usiamo “per decine” invece che “per milioni” e quindi dividiamo la somma per 10 invece che per 1.000.000.
Dividere i valori RPK per il fattore di scala “per milione”
Gene
Sample 1 TPM
Sample 2 TPM
Sample 3 TPM
A (2kb)
3.33
2.96
3.33
B (4kb)
3.33
3.09
3.33
C (1kb)
3.33
3.95
3.33
D (10kb)
0
0
0.1
A differenza di RPKM e FPKM, quando si calcola il TPM, si normalizza prima per la lunghezza del gene e poi per la profondità di sequenziamento. Tuttavia, gli effetti di questa differenza sono piuttosto profondi, come abbiamo già visto con l’esempio.
Le somme di ogni colonna sono molto diverse:
RPKM
Gene
Sample 1 RPKM
Sample 2 RPKM
Sample 3 RPKM
A (2kb)
1.43
1.33
1.42
B (4kb)
1.43
1.39
1.42
C (1kb)
1.43
1.78
1.42
D (10kb)
0
0
0.009
Total
4.29
4.5
4.25
TPM
Gene
Sample 1 TPM
Sample 2 TPM
Sample 3 TPM
A (2kb)
3.33
2.96
3.33
B (4kb)
3.33
3.09
3.33
C (1kb)
3.33
3.95
3.33
D (10kb)
0
0
0.1
Total
10
10
10
La somma di tutti i TPM in ogni campione è la stessa. In questo modo è più facile confrontare la proporzione di letture mappate su un gene in ciascun campione. Al contrario, con RPKM e FPKM, la somma delle letture normalizzate in ciascun campione può essere diversa e ciò rende più difficile il confronto diretto tra i campioni.
Nell’esempio, il TPM per il gene A nel campione 1 è 3,33 e nel campione 2 è 3,33. La stessa proporzione di letture totali mappate sul gene A in entrambi i campioni (0,33 in questo caso). In effetti, la somma dei TPM in entrambi i campioni è lo stesso numero (10 in questo caso), il denominatore necessario per calcolare le proporzioni è quindi lo stesso indipendentemente dal campione, e quindi la proporzione di letture per il gene A (3,33/10 = 0,33) per entrambi i campioni.
Con RPKM o FPKM, è più difficile confrontare la proporzione di letture totali perché la somma delle letture normalizzate in ogni campione può essere diversa (4,29 per il campione 1 e 4,25 per il campione 2). Pertanto, se l’RPKM per il gene A nel campione 1 è 1,43 e nel campione B è 1,43, non sappiamo se la stessa proporzione di letture nel campione 1 è mappata al gene A come nel campione 2.
Poiché l’RNA-Seq consiste nel confrontare la proporzione relativa di letture, il TPM sembra più appropriato di RPKM/FPKM.
L’RNA-Seq è spesso usato per confrontare un tipo di tessuto con un altro, per esempio, muscolo contro tessuto epiteliale. È possibile che vi siano molti geni specifici del muscolo trascritti nel muscolo ma non nel tessuto epiteliale. Si tratta di una differenza nella composizione della libreria.
È anche possibile osservare una differenza nella composizione della libreria nello stesso tipo di tessuto dopo l’eliminazione di un fattore di trascrizione.
Immaginiamo di avere i conteggi RNA-Seq di 2 campioni (stessa dimensione della libreria: 635 letture), per un genoma con 6 geni. I geni hanno la stessa espressione in entrambi i campioni, tranne uno: solo il campione 1 trascrive il gene D, a un livello elevato (563 letture). Poiché la dimensione della libreria è la stessa per entrambi i campioni, il campione 2 ha 563 letture in più da distribuire sui geni A, B, C, E ed F.
Gene
Sample 1
Sample 2
A
30
235
B
24
188
C
0
0
D
563
0
E
5
39
F
13
102
Total
635
635
Di conseguenza, il numero di letture per tutti i geni, ad eccezione dei geni C e D, è molto alto nel campione 2. Tuttavia, l’unico genio differenziato è quello che è stato sequenziato con un’incertezza elevata (cioè basi di bassa qualità). Ciononostante, l’unico gene differenzialmente espresso è il gene D.
TPM, RPKM o FPKM non trattano queste differenze nella composizione della libreria durante la normalizzazione, ma strumenti più complessi, come DESeq2, lo fanno.
DESeq2 (Love et al. 2014) è un ottimo strumento per trattare i dati RNA-seq ed eseguire analisi di espressione genica differenziale (DGE). Prende i file del conteggio delle letture da diversi campioni, li combina in una grande tabella (con i geni nelle righe e i campioni nelle colonne) e applica la normalizzazione per la profondità di sequenziamento e la composizione della libreria. Non è necessario tenere conto della normalizzazione della lunghezza del gene perché stiamo confrontando i conteggi tra gruppi di campioni per lo stesso gene.
Facciamo un esempio per illustrare come DESeq2 scala i diversi campioni:
Gene
Campione 1
Campione 2
Campione 3
A
0
10
4
B
2
6
12
C
33
55
200
L’obiettivo è calcolare un fattore di scala per ogni campione, che tenga conto della profondità di lettura e della composizione della libreria.
Prendere il log\(\_e\) di tutti i valori:
Gene
log(Campione 1)
log(Campione 2)
log(Campione 3)
A
-Inf
2,3
1,4
B
0,7
1,8
2,5
C
3,5
4,0
5,3
Media di ogni riga:
Gene
Media dei valori log
A
-Inf
B
1,7
C
4,3
La media dei valori logici (nota anche come media geometrica) viene utilizzata in questo caso perché non è facilmente influenzata da valori anomali (ad esempio, il gene C con il suo valore anomalo per il campione 3).
Filtrare i geni il cui valore è infinito.
Gene
Media dei valori log
B
1,7
C
4,3
Qui filtriamo i geni che non hanno conteggi di letture in almeno un campione, ad esempio i geni trascritti solo in un tessuto, come il gene D nell’esempio precedente. Questo aiuta a concentrare i fattori di scala sui geni trascritti a livelli simili, indipendentemente dalla condizione.
Sottrarre il valore medio del log dai conteggi del log:
Gene
log(Campione 1)
log(Campione 2)
log(Campione 3)
B
-1,0
0,1
0,8
C
-0,8
-0,3
1,0
\[log(\textrm{conteggio per il gene X}) - average(\textrm{valori logici per il conteggio per il gene X}) = log(\frac{\textrm{conteggio per il gene X}}{\textrm{media per il gene X}})\]
Questo passaggio confronta il rapporto tra i conteggi di ciascun campione e la media di tutti i campioni.
Calcolare la mediana dei rapporti per ciascun campione:
Gene
log(Campione 1)
log(Campione 2)
log(Campione 3)
B
-1,0
0,1
0,8
C
-0,8
-0,3
1,0
Mediana
-0,9
-0,1
0,9
La mediana viene utilizzata per evitare che i geni estremi (probabilmente rari) influenzino troppo il valore in una direzione. Aiuta a dare maggiore enfasi ai geni moderatamente espressi.
Calcolare il fattore di scala prendendo l’esponenziale delle mediane:
Gene
Campione 1
Campione 2
Campione 3
Mediana
-0,9
-0,1
0,9
Fattori di scala
0,4
0,9
2,5
Calcolo dei conteggi normalizzati: dividere i conteggi originali per i fattori di scala:
DESeq2 esegue anche l’analisi dell’espressione genica differenziale (DGE), che ha due compiti fondamentali:
Stimare la varianza biologica utilizzando le repliche per ogni condizione
Stimare la significatività delle differenze di espressione tra due condizioni qualsiasi
Questa analisi dell’espressione è stimata dal conteggio delle letture e si cerca di correggere la variabilità delle misurazioni usando i replicati, che sono assolutamente essenziali per ottenere risultati accurati. Per la vostra analisi, vi consigliamo di utilizzare almeno 3, ma preferibilmente 5 repliche biologiche per condizione. È possibile avere un numero diverso di repliche per condizione.
Un replicato tecnico è un esperimento che viene eseguito una volta ma misurato più volte (ad esempio, sequenziamento multiplo della stessa libreria). Un replicato biologico è un esperimento eseguito (e anche misurato) più volte.
Nei nostri dati, abbiamo 4 repliche biologiche (qui chiamate campioni) senza trattamento e 3 repliche biologiche con trattamento (gene Pasilla impoverito da RNAi).
Si consiglia di combinare le tabelle di conteggio per i diversi replicati tecnici (ma non per i replicati biologici) prima di un’analisi di espressione differenziale (cfr. documentazione DESeq2)
È possibile incorporare nell’analisi fattori multipli con diversi livelli che descrivono fonti note di variazione (ad esempio, trattamento, tipo di tessuto, sesso, lotti), con due o più livelli che rappresentano le condizioni per ciascun fattore. Dopo la normalizzazione è possibile confrontare la risposta dell’espressione di qualsiasi gene alla presenza di diversi livelli di un fattore in modo statisticamente affidabile.
Nel nostro esempio, abbiamo campioni con due fattori variabili che possono contribuire a differenze nell’espressione genica:
Trattamento (trattato o non trattato)
Tipo di sequenziamento (paired-end o single-end)
Qui il trattamento è il fattore principale che ci interessa. Il tipo di sequenziamento è un’ulteriore informazione che conosciamo sui dati e che potrebbe influenzare l’analisi. L’analisi multifattoriale ci permette di valutare l’effetto del trattamento, tenendo conto anche del tipo di sequenziamento.
Commento
Si consiglia di aggiungere tutti i fattori che si pensa possano influenzare l’espressione genica nel proprio esperimento. Può trattarsi del tipo di sequenziamento, come in questo caso, ma anche della manipolazione (se diverse persone sono coinvolte nella preparazione delle librerie), di altri effetti del batch, ecc.
Se avete solo uno o due fattori con un numero limitato di repliche biologiche, la configurazione di base di DESeq2 è sufficiente. Nel caso di un setup sperimentale complesso con un gran numero di repliche biologiche, sono appropriate le raccolte basate sui tag. Entrambi gli approcci danno gli stessi risultati. L’approccio basato sui tag richiede alcuni passaggi aggiuntivi prima di eseguire lo strumento DESeq2, ma si rivelerà vantaggioso quando si lavora con un setup sperimentale complesso.
Hands-on: Choose Your Own Tutorial
Questa è una sezione 'Choose Your Own Tutorial' (CYOT) (nota anche come 'Choose Your Own Analysis' (CYOA)), in cui è possibile scegliere tra più percorsi. Fare clic su uno dei pulsanti sottostanti per selezionare il modo in cui si desidera seguire l'esercitazione
Quale approccio preferisci usare?
Ora possiamo eseguire DESeq2:
Pratica: Determinare le caratteristiche differenzialmente espresse
DESeq2 ( Galaxy version 2.11.40.8+galaxy0) con i seguenti parametri:
“come “: Select datasets per level
In “Factor “:
“Specify a factor name, e.g. effects_drug_x or cancer_markers “: Treatment
In “1: Factor level”:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: treated’”*
In “FiCount file(s)”: Select the collection treated
In ”: Factor level”:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: untreated`
In “Count file(s)”: Select the collection untreated
param-repeat“Insert Factor”
“Specify a factor name, e.g. effects_drug_x or cancer_markers”: Sequencing
In “Factor level”:
param-repeat“Insert Factor level”
“pecify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: PE
In “Count file(s)”: Select the collection paired
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: SE
In “File di conteggio “: Count file(s)"*: Select the collection single`
“Files have header? “: Yes
“Choice of Input data”: Count data (e.g. from HTSeq-count, featureCounts or StringTie)
In “dvanced options”:
“Use beta priors”: Yes
In “Output options”:
“Output selector”: Generate plots for visualizing the analysis results, Output normalised counts
DESeq2 richiede di specificare, per ciascun fattore sperimentale, i conteggi dei campioni appartenenti a ogni categoria. Per questo motivo, useremo i tag sulla nostra raccolta di conteggi, così da poter selezionare facilmente tutti i campioni appartenenti alla stessa categoria.Per ulteriori informazioni su metodi alternativi per impostare i tag di gruppo, consultarequesto tutorial.
Pratica: Aggiungi dei tag alla tua collezione per ognuno di questi fattori
Creare una raccolta di dataset contenente tutti i file di conteggio e assegnarle il nome all counts. Nominare ogni elemento in modo che contenga solo l’id GSM, il trattamento e la libreria, ad esempio, GSM461176_untreat_single.
Cliccare su galaxy-selectorSeleziona elementi nella parte superiore del pannello della cronologia
Controllare tutti i dataset della cronologia che si desidera includere
Fare clic su n di N selezionati e scegliere Costruisci elenco dataset
Inserire un nome per la raccolta
Fare clic su Crea raccolta per creare la raccolta
Fare di nuovo clic sull’icona del segno di spunta in cima alla cronologia
Extract element identifiers ( Galaxy version 0.0.2) con i seguenti parametri:
param-collection“Dataset collection”: all counts
Ora estrarremo dai nomi i fattori:
Replace Text in entire line ( Galaxy version 9.3+galaxy1)
param-file“File to process”: output di Extract element identifierstool
In “Replacement “:
In “1: Replacement “
“Find pattern”: (.*)_(.*)_(.*)
“Replace with”: \1_\2_\3\tgroup:\2\tgroup:\3
Questo passaggio crea 2 colonne aggiuntive con il tipo di trattamento e di sequenziamento che possono essere utilizzate con lo strumento Elementi tag
Cambiare il tipo di dati in tabular
Cliccare sull’icona galaxy-pencilicona della matita per il set di dati per modificarne gli attributi
Nel pannello centrale, fare clic su galaxy-chart-select-data *scheda *Datipi** in alto
Nella sezione galaxy-chart-select-dataAssegna tipo di dato, selezionare tabular dal menu a discesa “Nuovo tipo”
Suggerimento: si può iniziare a digitare il tipo di dato nel campo per filtrare il menu a discesa
Fare clic sul pulsante Salva
Tag elements
param-collection“Collezione di input “: all counts
param-file“Tagga gli elementi della raccolta in base a questo file “: output di Replace Texttool
Ispezione della nuova raccolta
A prima vista non si nota perché i nomi sono gli stessi. Tuttavia, se si fa clic su uno di essi e si clicca su galaxy-tagsEdit dataset tags, si dovrebbero vedere 2 tag che iniziano con “group:”. Questa parola chiave permetterà di usare questi tag in DESeq2.
Ora possiamo eseguire DESeq2:
Pratica: Determinare le caratteristiche differenzialmente espresse
DESeq2 ( Galaxy version 2.11.40.8+galaxy0) with the following parameters:
“how”: Select group tags corresponding to levels
param-collection“Count file(s) collection”: output of Tag elementstool
In “Factor”:
param-repeat“Insert Factor”
“Specify a factor name, e.g. effects_drug_x or cancer_markers”: Treatment
In “Factor level”:
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: treated
“Select groups that correspond to this factor level”: Tags: treat
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: untreated
“Select groups that correspond to this factor level”: Tags: untreat
param-repeat“Insert Factor”
“Specify a factor name, e.g. effects_drug_x or cancer_markers”: Sequencing
In “Factor level”:
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: PE
“Select groups that correspond to this factor level”: Tags: paired
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: SE
“Select groups that correspond to this factor level”: Tags: single
“Files have header?”: Yes
“Choice of Input data”: Count data (e.g. from HTSeq-count, featureCounts or StringTie)
In “Advanced options”:
“Use beta priors”: Yes
In “Output options”:
“Output selector”: Generate plots for visualizing the analysis results, Output normalised counts
DESeq2 richiede di fornire per ogni fattore, i conteggi dei campioni in ogni categoria. Utilizzeremo quindi dei pattern sul nome dei nostri campioni per selezionare facilmente tutti i campioni appartenenti alla stessa categoria.
Pratica: Generare una raccolta di ogni categoria
Creare una raccolta di dataset contenente tutti i file di conteggio e assegnarle il nome all counts. Rinominare ciascun elemento della raccolta in modo che includa solo l’ID GSM, la condizione di trattamento e il tipo di libreria, ad esempio GSM461176_untreat_single.
Cliccare su galaxy-selectorSeleziona elementi nella parte superiore del pannello della cronologia
Controllare tutti i dataset della cronologia che si desidera includere
Fare clic su n di N selezionati e scegliere Costruisci elenco dataset
Inserire un nome per la raccolta
Fare clic su Crea raccolta per creare la raccolta
Fare di nuovo clic sull’icona del segno di spunta in cima alla cronologia
Extract element identifiers ( Galaxy version 0.0.2) con i seguenti parametri:
param-collection“Dataset collection “: all counts
Ora divideremo la raccolta per trattamento. Dobbiamo trovare un modello che sia presente in una sola delle due categorie. Utilizzeremo la parola untreat:
Search in textfiles ( Galaxy version 9.3+galaxy1) (grep) with the following parameters:
“Select lines from”: Extract element identifiers on data XXX (output of Extract element identifierstool)
“that”: Match
“Regular Expression”: untreat
Filter collecion con i seguenti parametri:
“Input collection”: all counts
“How should the elements to remove be determined”: Remove if identifiers are ABSENT from file
“Filter out identifiers absent from”: Search in textfiles on data XXX (output of Search in textfilestool)
Rinominare entrambe le raccolte untreated (la raccolta filtrata) e treated (la raccolta scartata).
Ripeteremo lo stesso processo usando single
Search in textfiles ( Galaxy version 9.3+galaxy1) (grep) with the following parameters:
“Select lines from”: Extract element identifiers on data XXX (output of Extract element identifierstool)
“that”: Match
“Regular Expression”: single
Filter collecion con i seguenti parametri:
“Input collection”: all counts
“How should the elements to remove be determined”: Remove if identifiers are ABSENT from file
“Filter out identifiers absent from”: Search in textfiles on data XXX (output of Search in textfilestool)
Rinominare entrambe le raccolte single (la raccolta filtrata) e paired (la raccolta scartata).
Ora possiamo eseguire DESeq2:
Pratica: Determinare le caratteristiche differenzialmente espresse
DESeq2 ( Galaxy version 2.11.40.8+galaxy0) with the following parameters:
“how”: Select datasets per level
In “Factor”:
“Specify a factor name, e.g. effects_drug_x or cancer_markers”: Treatment
In “1: Factor level”:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: treated
param-collection“Count file(s)”: Select the collection treated
In “2: Factor level”:
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: untreated
param-collection“Count file(s)”: Select the collection untreated
param-repeat“Insert Factor”
“Specify a factor name, e.g. effects_drug_x or cancer_markers”: Sequencing
In “Factor level”:
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: PE
param-collection“Count file(s)”: Select the collection paired
param-repeat“Insert Factor level”
“Specify a factor level, typical values could be ‘tumor’, ‘normal’, ‘treated’ or ‘control’“: SE
param-collection“Count file(s)”: Select the collection single
“Files have header?”: Yes
“Choice of Input data”: Count data (e.g. from HTSeq-count, featureCounts or StringTie)
In “Advanced options”:
“Use beta priors”: Yes
In “Output options”:
“Output selector”: Generate plots for visualizing the analysis results, Output normalised counts
DESeq2 ha generato 3 risultati:
Una tabella con i conteggi normalizzati per ciascun gene (righe) nei campioni (colonne)
Un riassunto grafico dei risultati, utile per valutare la qualità dell’esperimento:
Un grafico delle prime 2 dimensioni di un’analisi delle componenti principali (PCA), eseguita sui conteggi normalizzati dei campioni
Immaginiamo di avere alcune bottiglie di birra sul tavolo. Possiamo descrivere ogni birra in base al suo colore, alla sua schiuma, a quanto è forte e così via. Possiamo comporre un intero elenco di caratteristiche diverse di ogni birra in un negozio di birra. Ma molte di esse misureranno proprietà correlate e quindi saranno ridondanti. Se così fosse, dovremmo essere in grado di riassumere ogni birra con un numero minore di caratteristiche. Questo è ciò che fa la PCA o analisi delle componenti principali.
Con la PCA non ci limitiamo a selezionare alcune caratteristiche interessanti e a scartare le altre. Costruiamo invece alcune nuove caratteristiche che riassumono bene il nostro elenco di birre. Queste nuove caratteristiche sono costruite utilizzando quelle vecchie. Ad esempio, si può calcolare una nuova caratteristica, ad esempio la dimensione della schiuma meno il pH della birra. Si tratta di combinazioni lineari.
In effetti, la PCA trova le migliori caratteristiche possibili, quelle che riassumono l’elenco delle birre. Queste caratteristiche possono essere utilizzate per trovare somiglianze tra le birre e raggrupparle.
Tornando ai conteggi delle letture, la PCA viene eseguita sui conteggi normalizzati per tutti i campioni. Qui vogliamo descrivere i campioni in base all’espressione dei geni. Le caratteristiche sono quindi il numero di letture mappate su ciascun gene. Le utilizziamo e le loro combinazioni lineari per rappresentare i campioni e le loro somiglianze.
Mostra i campioni in un piano bidimensionale definito dalle prime due componenti principali.
Ogni replicato è rappresentato come un singolo punto dati.
Questo tipo di grafico è utile per visualizzare l’effetto complessivo delle variabili sperimentali e degli eventuali effetti di batch.
Figura 38: Principal component plot of the samples
Cosa separa la prima dimensione (PC1)?
E la seconda dimensione (PC2)?
Cosa possiamo concludere riguardo al design di DESeq (fattori e livelli) che abbiamo scelto?
La prima dimensione separa i campioni trattati da quelli non trattati.
La seconda dimensione separa i dataset single-end da quelli paired-end.
II dataset sono raggruppati in base ai livelli dei due fattori. Non sembra esserci alcun effetto nascosto nei dati. Se è presente una variazione indesiderata (ad esempio effetti di batch), è sempre consigliato correggerla, cosa che può essere fatta in DESeq2 includendo nel design eventuali variabili di batch note.
Heatmap della matrice delle distanze campione-campione (con clustering) basata sui conteggi normalizzati.
La heatmap fornisce una panoramica delle somiglianze e delle differenze tra i campioni: il colore rappresenta la distanza tra i campioni. Il blu scuro indica una distanza minore, cioè campioni più simili in base ai conteggi normalizzati
Figura 39: Heatmap delle distanze campione-campione
Come sono raggruppati i campioni?
Vengono prima raggruppate in base al trattamento (il primo fattore) e poi in base al tipo di sequenziamento (il secondo fattore), come nel grafico PCA.
Stime di dispersione: stime per gene (nero), valori adattati (rosso) e stime finali a posteriori massime utilizzate nei test (blu)
Questo grafico di dispersione è tipico: le stime finali vengono rimpicciolite (shrunken) dalle stime gene-wise verso le stime fitted. Alcune stime gene-wise sono contrassegnate come outlier e non vengono rimpicciolite verso il valore adattato.
L’entità del restringimento può essere maggiore o minore di quella mostrata qui, a seconda della dimensione del campione, del numero di coefficienti, della media delle righe e della variabilità delle stime gene-wise.
Istogramma dei valori di p per i geni nel confronto tra i 2 livelli del 1° fattore
Questo grafico mostra una visione globale della relazione tra la variazione di espressione tra le condizioni (log fold change, M), l’intensità media di espressione dei geni (A) e la capacità dell’algoritmo di rilevare l’espressione genica differenziale. I geni che superano la soglia di significatività (p-value aggiustato < 0,1) sono evidenziati in rosso.
Un file di riepilogo con i seguenti valori per ciascun gene:
Identificatori dei geni
Media dei conteggi normalizzati, calcolata su tutti i campioni di entrambe le condizioni
Variazione di espressione in log₂ (log₂ fold change), cioè il logaritmo in base 2 del rapporto tra le medie di espressione nelle due condizioni
I cambiamenti di fold log₂ si basano sul confronto tra il livello 1 e il livello 2 del fattore primario, pertanto l’ordine di inserimento dei livelli del fattore è importante. In questo caso, DESeq2 calcola le variazioni di espressione (log₂ fold change) dei campioni trattati rispetto a quelli non trattati per il primo fattore “Trattamento”. In altre parole, i valori ottenuti rappresentano l’aumento o la diminuzione dell’espressione genica nei campioni trattati rispetto ai non trattati.
Stima dell’errore standard per la stima del cambiamento log2 fold
valore p per la significatività statistica di questo cambiamento
valore p aggiustato per i test multipli con la procedura di Benjamini-Hochberg, che controlla il tasso di scoperta dei falsi (FDR)
Il valore p è una misura spesso utilizzata per determinare se una particolare osservazione possiede o meno una significatività statistica. In senso stretto, il valore p è la probabilità che i dati possano essere emersi casualmente, assumendo che l’ipotesi nulla sia corretta. Nel caso concreto di RNA-Seq, l’ipotesi nulla è che non vi sia espressione genica differenziale. Quindi un valore p di 0,13 per un particolare gene indica che, per quel gene, supponendo che non sia espresso in modo differenziato, c’è una probabilità del 13% che qualsiasi apparente espressione differenziale possa essere semplicemente prodotta da una variazione casuale nei dati sperimentali.
il 13% è ancora piuttosto alto, quindi non possiamo essere sicuri che sia in atto un’espressione genica differenziale. Il modo più comune in cui gli scienziati utilizzano i valori di p è quello di fissare una soglia (di solito 0,05, a volte altri valori come 0,01) e rifiutare l’ipotesi nulla solo per valori di p inferiori a questo valore. In questo modo, per i geni con valori di p inferiori a 0,05, possiamo affermare con sicurezza che l’espressione genica differenziale svolge un ruolo. Va notato che tale soglia è arbitraria e non c’è alcuna differenza significativa tra un valore di p di 0,049 e 0,051, anche se rifiutiamo l’ipotesi nulla solo nel primo caso.
Sfortunatamente, i valori p sono spesso utilizzati in modo improprio nella ricerca scientifica, tanto che Wikipedia offre un articolo dedicato sull’argomento. Si veda anche questo articolo (rivolto a un pubblico generico e non scientifico).
Per ulteriori informazioni su DESeq2 e i suoi risultati, è possibile consultare la documentazione di DESeq2.
Domanda
Il gene FBgn0003360 è differenzialmente espresso a causa del trattamento? Se sì, di quanto?
Il gene Pasilla (ps, FBgn0261552) è downregolato dal trattamento RNAi?
Potremmo anche essere ipoteticamente interessati all’effetto del sequenziamento (o di altri fattori secondari in altri casi). Come potremmo conoscere i geni espressi in modo differenziato a causa del tipo di sequenziamento?
Vorremmo analizzare l’interazione tra il trattamento e il sequenziamento. Come possiamo farlo?
FBgn0003360 è differenzialmente espresso a causa del trattamento: ha un valore p significativo aggiustato (\(2,8 \cdot 10^{-171} << 0,05\)). È meno espresso (- nella colonna log2FC) nei campioni trattati rispetto a quelli non trattati, di un fattore ~8 (\(2^{log2FC} = 2^{2.99542318410271}\)).
È possibile verificare manualmente la presenza di FBgn0261552 nella prima colonna o eseguire Filtrare i dati su qualsiasi colonna usando espressioni semplici
param-file“Filtro “: il DESeq2 result file (output di DESeq2tool)
“Con le seguenti condizioni “: c1 == "FBgn0261552"
Il log2 fold-change è negativo, quindi è effettivamente downregolato e il valore p aggiustato è inferiore a 0,05, quindi fa parte dei geni significativamente modificati.
DESeq2 in Galaxy restituisce il confronto tra i diversi livelli per il 1° fattore, dopo la correzione per la variabilità dovuta al 2° fattore. Nel nostro caso attuale, trattati contro non trattati per qualsiasi tipo di sequenziamento. Per confrontare i tipi di sequenziamento, dovremmo eseguire nuovamente DESeq2 cambiando i fattori: il fattore 1 (trattamento) diventa fattore 2 e il fattore 2 (sequenziamento) diventa fattore 1.
Per aggiungere l’interazione tra due fattori (ad es. trattati per i dati paired-end vs. non trattati per quelli single-end), dovremmo eseguire DESeq2 un’altra volta ma con un solo fattore con i seguenti 4 livelli:
trattato-PE
non trattato-PE
trattato-SE
non trattato-SE
Selezionando “Output all levels vs all levels of primary factor (use when you have >2 levels for primary factor) “ a Yes, possiamo confrontare treated-PE vs untreated-SE.
Annotazione dei risultati DESeq2
L’ID di ogni gene è qualcosa come FBgn0003360, che è un ID del database corrispondente, in questo caso Flybase (Thurmond et al. 2018). Questi ID sono univoci, ma a volte preferiamo avere i nomi dei geni, anche se potrebbero non fare riferimento a un gene unico (ad esempio, duplicato dopo una ri-annotazione). Ma i nomi dei geni possono già suggerire una funzione o aiutare a cercare i candidati desiderati. Vorremmo anche visualizzare la posizione di questi geni all’interno del genoma. Possiamo estrarre tali informazioni dal file di annotazione utilizzato per la mappatura e il conteggio.
Pratica: Annotazione dei risultati DESeq2
Importare l’annotazione genica Ensembl per Drosophila melanogaster (Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz) dalla storia precedente, o dalla libreria Shared Data o da Zenodo:
Annotate DESeq2/DEXSeq output tables ( Galaxy version 1.1.0) with:
param-file“Tabular output of DESeq2/edgeR/limma/DEXSeq”: the DESeq2 result file (output of DESeq2tool)
“Input file type”: DESeq2/edgeR/limma
param-file“Reference annotation in GFF/GTF format”: imported gtf Drosophila_melanogaster.BDGP6.32.109_UCSC.gtf.gz
L’output generato è un’estensione del file precedente:
Identificatori dei geni
Conteggi medi normalizzati su tutti i campioni
Log2 fold change (cambiamento di ripiegamento)
Stima dell’errore standard per la stima del cambiamento log2 fold
Statistica di Wald
valore p per la statistica di Wald
valore p aggiustato per i test multipli con la procedura Benjamini-Hochberg per la statistica di Wald
Cromosoma
Inizio
Fine
Filamento
Caratteristica
Nome del gene
Domanda
Dove si trova il gene più sovraespresso?
Qual è il nome del gene?
Dove si trova il gene Pasilla (FBgn0261552)?
FBgn0025111 (il gene in cima alla classifica con il valore log2FC positivo più alto) si trova sul filamento inverso del cromosoma X, tra 10.778.953 bp e 10.786.907 bp.
Dalla tabella, abbiamo ottenuto il simbolo del gene: Ant2. Dopo una ricerca su online biological databases, scopriamo che Ant2 corrisponde all’adenina nucleotide translocase 2.
Il gene Pasilla si trova sul filamento anteriore del cromosoma 3R, tra 9.417.939 bp e 9.455.500 bp.
La tabella annotata non contiene nomi di colonne, il che rende difficile la lettura. Vorremmo aggiungerli prima di andare avanti.
Pratica: Aggiungi nomi di colonne
Creare un nuovo file (header) da quanto segue (riga d’intestazione dell’output di DESeq2)
GeneID Base mean log2(FC) StdErr Wald-Stats P-value P-adj Chromosome Start End Strand Feature Gene name
Fare clic su galaxy-uploadCaricamento dati nella parte superiore del pannello degli strumenti
Selezionare galaxy-wf-editIncolla/Recupera dati in basso
Incollare il contenuto del file nel campo di testo
Cambiare il nome del dataset da “New File” a header * Cambiare Type da “Auto-detect” a tabular* Premere Avvio e Chiudere la finestra
Concatenate multiple datasets or collections per aggiungere questa riga di intestazione all’output Annotate:
param-file“Concatenate Dataset”: the header dataset
“Dataset”
Click on param-repeat“Insert Dataset”
param-file“select”: output of Annotatetool
Rinominare l’output in Annotated DESeq2 results
Estrazione e annotazione dei geni espressi in modo differenziato
Ora vorremmo estrarre i geni più differenzialmente espressi dal trattamento con una variazione > 2 (o < 1/2).
Pratica: Estrarre i geni con la maggiore espressione differenziale
Filtra i dati su qualsiasi colonna usando espressioni semplici per estrarre i geni con un cambiamento significativo nell’espressione genica (valore p aggiustato inferiore a 0,05) tra i campioni trattati e non trattati:
param-file“Filter”: Annotated DESeq2 results
“With following condition”: c7<0.05
“Number of header lines to skip”: 1
Rinominare l’output Genes with significant adj p-value.
Domanda
Quanti geni presentano un cambiamento significativo nell’espressione genica tra queste condizioni?
Otteniamo 966 (967 righe inclusa l’intestazione) geni (4,04%) con un cambiamento significativo nell’espressione genica tra campioni trattati e non trattati.
Commento
Il file con i risultati filtrati in modo indipendente può essere utilizzato per ulteriori analisi a valle, poiché esclude i geni con solo pochi conteggi di letture, in quanto questi geni non saranno considerati significativamente espressi in modo differenziato.
Selezioneremo ora solo i geni con un fold change (FC) > 2 o FC < 0,5. Si noti che il file di output di DESeq2 contiene \(log\_{2} FC\), piuttosto che FC stesso, quindi filtriamo per \(abs(log\_{2} FC) > 1\) (che implica FC > 2 o FC < 0,5).
Filtra i dati su qualsiasi colonna usando espressioni semplici per estrarre i geni con un \(abs(log\_{2} FC) > 1\):
param-file“Filter”: Genes with significant adj p-value
È possibile trovare il gene Pasilla (ps, FBgn0261552) in questa tabella?
Otteniamo 113 geni (114 righe compresa l’intestazione), ovvero l’11,79% dei geni significativamente espressi in modo differenziato.
Il gene Pasilla può essere trovato con una rapida ricerca (o anche usando Filtra i dati su qualsiasi colonna usando semplici espressioni)
Ora abbiamo una tabella con 113 righe e un’intestazione corrispondente ai geni più differenzialmente espressi. Per ogni gene abbiamo il suo ID, la media dei conteggi normalizzati (mediati su tutti i campioni di entrambe le condizioni), il suo \(log\_{2} FC\) e altre informazioni, tra cui il nome e la posizione del gene.
Visualizzazione dell’espressione dei geni differenzialmente espressi
Potremmo tracciare il \(log\_{2} FC\) per i geni estratti, ma qui vorremmo osservare una heatmap dell’espressione di questi geni nei diversi campioni. Dobbiamo quindi estrarre i conteggi normalizzati per questi geni.
Procediamo in più fasi:
Estrarre e tracciare i conteggi normalizzati per questi geni per ciascun campione con una heatmap, utilizzando il file dei conteggi normalizzati generato da DESeq2
Calcolare, estrarre e tracciare il punteggio Z dei conteggi normalizzati
Commento: Tutorial avanzato sulla visualizzazione
In questa esercitazione, spiegheremo rapidamente alcune possibili visualizzazioni. Per maggiori dettagli, consultare le esercitazioni aggiuntive sulla visualizzazione dei risultati di RNA-Seq:
Per estrarre i conteggi normalizzati per i geni interessanti, uniamo la tabella dei conteggi normalizzati generata da DESeq2 con la tabella appena generata. In questo modo manterremo solo le colonne corrispondenti ai conteggi normalizzati.
Pratica: Estrarre i conteggi normalizzati dei geni più differenzialmente espressi
Join two Datasets side by side on a specified field con i seguenti parametri:
param-file“Join”: the Normalized counts file (output of DESeq2tool)
“using column”: Column: 1
param-file“with”: Genes with significant adj p-value & abs(log2(FC)) > 1
“and column”: Column: 1
“Keep lines of first input that do not join with second input”: No
“Keep the header lines”: Yes
Il file generato ha più colonne di quelle necessarie per la mappa di calore: conteggi medi normalizzati, \(log\_{2} FC\) e altre informazioni di annotazione. Dobbiamo rimuovere le colonne extra.
Cut colonne da una tabella con i seguenti parametri per estrarre le colonne con gli ID dei geni e i conteggi normalizzati:
“Cut columns”: c1-c8
“Delimited by”: Tab
param-file“From”: the joined dataset (output of Join two Datasetstool)
Rinominare l’output in Normalized counts for the most differentially expressed genes
Ora abbiamo una tabella con 114 righe (i 113 geni più differenzialmente espressi e un’intestazione) e i conteggi normalizzati per questi geni nei 7 campioni.
Pratica: Plottare la mappa di calore dei conteggi normalizzati di questi geni per i campioni
heatmap2 ( Galaxy version 3.1.3.1+galaxy0) per tracciare la heatmap:
param-file“Input should have column headers”: Normalized counts for the most differentially expressed genes
“Data transformation”: Log2(value+1) transform my data
“Enable data clustering”: Yes
“Labeling columns and rows”: Label columns and not rows
Figura 40: Conteggi normalizzati per i geni più differenzialmente espressi
Domanda
Cosa rappresenta l’asse X della heatmap? E l’asse Y?
Si osserva qualcosa nel raggruppamento dei campioni e dei geni?
Cosa cambia se si rigenera la heatmap, questa volta selezionando Plot the data as it is in “Trasformazione dei dati “?
Perché non possiamo usare Log2(value) transform my data in “Trasformazione dei dati “?
Come si può generare una heatmap dei conteggi normalizzati per tutti i geni up-regulated con fold change > 2?
L’asse X mostra i 7 campioni, insieme a un dendrogramma che rappresenta la somiglianza tra i loro livelli di espressione genica. L’asse Y mostra i 113 geni differenzialmente espressi, insieme a un dendrogramma che rappresenta la somiglianza tra i livelli di espressione genica.
I campioni sono raggruppati per trattamento.
La scala cambia e si vedono solo pochi geni.
Perché l’espressione normalizzata del gene FBgn0013688 in GSM461180_treat_paired è a 0.
Estrarre i geni con \(log\_{2} FC\) > 1 (filtro per i geni con c3>1 nel riepilogo dei geni differenzialmente espressi) ed eseguire heatmap2tool sulla tabella generata.
Visualizzazione del punteggio Z
Per confrontare l’espressione genica tra i campioni, si può usare anche il punteggio Z, spesso rappresentato nelle pubblicazioni.
Il punteggio Z indica il numero di deviazioni standard che un valore ha rispetto alla media di tutti i valori dello stesso gruppo, in questo caso lo stesso gene. Un punteggio Z di -2 per il gene X nel campione A significa che questo valore è inferiore di 2 deviazioni standard rispetto alla media dei valori del gene X in tutti i campioni (A, B, C, ecc.).
Lo Z-score \(z\_{i,j}$ per un gene\)i\(in un campione\)j\(dato il conteggio normalizzato\)x_{i,j}$ è calcolato come \(z\_{i,j}$ = \frac{x\_{i,j}- \overline{x\_i}}{s\_i}\) con \(\overline{x\_i}\) la media e \(s\_i\) la deviazione standard dei conteggi normalizzati per il gene \(i\) su tutti i campioni.
Spesso abbiamo bisogno del punteggio Z per alcune visualizzazioni. Per calcolare il punteggio Z, il processo viene suddiviso in due fasi:
Sottrarre ogni valore per la media dei valori della riga (cioè \(x\_{i,j}- \overline{x\_i}\)) utilizzando la tabella di conteggio normalizzata
Dividere i valori precedenti per la deviazione standard dei valori di riga, usando 2 tabelle (i conteggi normalizzati e la tabella calcolata nel passaggio precedente)
Pratica: Calcolo del punteggio Z di tutti i geni
Table Compute ( Galaxy version 1.2.4+galaxy0) con i seguenti parametri per > sottrarre innanzitutto i valori medi per riga
“Input Single or Multiple Tables”: Single Table
param-file“Table”: Normalized counts file on data ... and others (output of DESeq2tool)
“Type of table operation”: Perform a full table operation
“Operation”: Custom
“Custom expression on ‘table’, along ‘axis’ (0 or 1)”: table.sub(table.mean(1), 0)`
L’espressione table.mean(1) calcola la media per ogni riga (qui i geni) e table.sub(table.mean(1), 0) sottrae ogni valore dalla media della riga (calcolata con table.mean(1))
Table Compute ( Galaxy version 1.2.4+galaxy0) con i seguenti parametri:
“Input Single or Multiple Tables”: Multiple Table
Click on param-repeat“Insert Tables”
In “1: Tables”:
param-file“Table”: Normalized counts file on data ... and others (output of DESeq2tool)
Click on param-repeat“Insert Tables”
In “2: Tables”:
param-file“Table”: output of the first Table Computetool
“Custom expression on ‘tableN’“: table2.div(table1.std(1),0)
L’espressione table1.std(1) calcola le deviazioni standard di ogni riga della prima tabella (conteggi normalizzati) e table2.div divide i valori della seconda tabella (precedentemente calcolati) per queste deviazioni standard.
Rinominare l’output in Z-scores
Ispezione del file di output
Ora abbiamo una tabella con il punteggio Z per tutti i geni nei 7 campioni.
Domanda
Qual è l’intervallo per il punteggio Z?
Perché alcune righe sono vuote?
Cosa possiamo dire dei punteggi Z per i geni differenzialmente espressi (ad esempio, FBgn0037223)?
Possiamo usare il punteggio Z per stimare la forza dell’espressione differenziale di un gene?
Il punteggio Z varia da -3 deviazioni standard a +3 deviazioni standard. Può essere posizionato su una curva di distribuzione normale: -3 è l’estrema sinistra della curva di distribuzione normale e +3 l’estrema destra della curva di distribuzione normale
Se tutti i conteggi sono identici (di solito a 0), la deviazione standard è 0, il punteggio Z non può essere calcolato per questi geni.
Quando un gene è espresso in modo differenziato tra due gruppi (qui trattati e non trattati), i punteggi Z per questo gene saranno (per lo più) positivi per i campioni di un gruppo e (per lo più) negativi per i campioni dell’altro gruppo.
Il punteggio Z è un rapporto segnale/rumore. Grandi punteggi Z assoluti, cioè grandi valori positivi o negativi, non sono una stima diretta dell’effetto, cioè della forza dell’espressione differenziale. Uno stesso Z-score grande può avere significati diversi, a seconda del rumore:
con un grande rumore: un effetto molto grande
con un po’ di rumore: un effetto piuttosto ampio
con solo poco rumore: un effetto piuttosto piccolo
con rumore quasi nullo: un effetto minuscolo
Il problema è che il “rumore” in questo caso non è solo il rumore della misura. Può anche essere legato alla “rigidità” del controllo della regolazione genica. I geni non strettamente controllati, cioè la cui espressione può variare in un ampio intervallo di campioni, possono essere notevolmente indotti o repressi. Il loro Z-score assoluto sarà piccolo perché le variazioni tra i campioni sono grandi. Al contrario, i geni strettamente controllati possono subire solo cambiamenti molto piccoli nella loro espressione, senza alcun impatto biologico. Per questi geni il punteggio Z assoluto sarà grande.
Ora vorremmo tracciare una heatmap per i punteggi Z:
Figura 41: Z-scores for the most differentially expressed genes
Pratica: Plottare lo Z-score dei geni più differenzialmente espressi
heatmap2 ( Galaxy version 3.1.3.1+galaxy0) per tracciare la heatmap:
param-file“Input should have column headers”: Normalized counts for the most differentially expressed genes
“Data transformation”: Plot the data as it is
“Compute z-scores prior to clustering”: Compute on rows
“Enable data clustering”: Yes
“Labeling columns and rows”: Label columns and not rows
“Type of colormap to use”: Gradient with 3 colors
Analisi dell’arricchimento funzionale dei geni DE
Abbiamo estratto i geni che sono espressi in modo differenziato nei campioni trattati (privi di geni PS) rispetto a quelli non trattati. Ora, vorremmo sapere se i geni espressi in modo differenziato sono trascritti in modo arricchito da geni che appartengono a categorie più comuni o specifiche, al fine di identificare le funzioni biologiche che potrebbero essere influenzate.
Analisi dell’ontologia del gene
L’analisi Gene Ontology (GO) è ampiamente utilizzata per ridurre la complessità ed evidenziare i processi biologici negli studi di espressione genomica. Tuttavia, i metodi standard forniscono risultati distorti sui dati RNA-Seq a causa dell’eccessiva individuazione dell’espressione differenziale per i trascritti lunghi e altamente espressi.
goseq (Young et al. 2010) fornisce metodi per eseguire l’analisi GO dei dati RNA-Seq tenendo conto del bias di lunghezza. goseq potrebbe essere applicato anche ad altre analisi dei dati RNA-Seq basate su categorie, come l’analisi dei percorsi KEGG, come discusso in un’altra sezione.
goseq ha bisogno di 2 file come input:
Un file tabellare con i geni differenzialmente espressi di tutti i geni analizzati nell’esperimento RNA-Seq con 2 colonne:
gli ID dei geni (unici all’interno del file), in lettere maiuscole
un booleano che indica se il gene è differenzialmente espresso o meno (True se differenzialmente espresso o False se no)
Un file con informazioni sulla lunghezza di un gene per correggere i potenziali bias di lunghezza nei geni differenzialmente espressi
Pratica: Preparare il primo set di dati per goseq
Compute ( Galaxy version 2.0) su righe con i seguenti parametri:
param-file“Input file”: the DESeq2 result file (output of DESeq2tool)
In “Expressions”:
param-text“Add expression”: bool(float(c7)<0.05)
param-select“Mode of the operation?”: Append
Under “Error handling”:
param-toggle“Autodetect column types”: No
param-select“If an expression cannot be computed for a row”: Fill in a replacement value
param-select“Replacement value”: `False
Cut colonne da una tabella con i seguenti parametri:
“Cut columns”: c1,c8
“Delimited by”: Tab
param-file“From”: the output of the Computetool
Change Case con
param-file“From”: the output of the previous Cuttool
“Change case of columns”: c1
“Delimited by”: Tab
“To”: Upper case
Rinominare l’output in Gene IDs and differential expression
Abbiamo appena generato il primo input per goseq. Come secondo input per goseq abbiamo bisogno delle lunghezze dei geni. Possiamo usare le lunghezze dei geni generate da featureCounts o Gene length and GC content e formattare gli ID dei geni.
Pratica: Preparare il file della lunghezza dei geni
Copiare la raccolta delle lunghezze delle caratteristiche precedentemente generata da featureCountstool in questa history
Esistono 3 modi per copiare i set di dati tra le cronologie
Dalla cronologia originale
Cliccare sull’icona galaxy-gear che si trova in cima all’elenco dei dataset nel pannello della cronologia
Fare clic su Copia set di dati
Selezionare i file desiderati
Dare un nome rilevante alla “Nuova cronologia”
Convalida per ‘Copia elementi della cronologia’
Fare clic sul nome della nuova cronologia nel riquadro verde appena visualizzato per passare a questa cronologia
Utilizzo dell’icona galaxy-columnsMostra le cronologie affiancate
Fare clic sulla freccia a discesa galaxy-dropdown in alto a destra del pannello della cronologia (opzioni cronologia)
Cliccare su galaxy-columnsMostra le cronologie affiancate
se la cronologia di destinazione non è presente
Fare clic su “Seleziona cronologia”
Fare clic sulla cronologia di destinazione
Convalidare con ‘Cambia selezionato’
Trascinare il dataset da copiare dalla sua cronologia originale
rilasciarlo nella cronologia di destinazione
Dalla cronologia di destinazione
Cliccare su Utente nella barra in alto
Cliccare su Datasets
Ricerca del set di dati da copiare
Fare clic sul suo nome
Cliccare su Copia nella cronologia corrente
Extract Dataset con:
param-file“From”: GSM461177_untreat_paired (output of Extract Datasettool)Gene length
“Change case of columns”: c1
“Delimited by”: Tab
“To”: Upper case
Copiare l’output di Gene length and GC contenttool (Gene length) in questa history
Esistono 3 modi per copiare i set di dati tra le cronologie
Dalla cronologia originale
Cliccare sull’icona galaxy-gear che si trova in cima all’elenco dei dataset nel pannello della cronologia
Fare clic su Copia set di dati
Selezionare i file desiderati
Dare un nome rilevante alla “Nuova cronologia”
Convalida per ‘Copia elementi della cronologia’
Fare clic sul nome della nuova cronologia nel riquadro verde appena visualizzato per passare a questa cronologia
Utilizzo dell’icona galaxy-columnsMostra le cronologie affiancate
Fare clic sulla freccia a discesa galaxy-dropdown in alto a destra del pannello della cronologia (opzioni cronologia)
Cliccare su galaxy-columnsMostra le cronologie affiancate
se la cronologia di destinazione non è presente
Fare clic su “Seleziona cronologia”
Fare clic sulla cronologia di destinazione
Convalidare con ‘Cambia selezionato’
Trascinare il dataset da copiare dalla sua cronologia originale
rilasciarlo nella cronologia di destinazione
Dalla cronologia di destinazione
Cliccare su Utente nella barra in alto
Cliccare su Datasets
Ricerca del set di dati da copiare
Fare clic sul suo nome
Cliccare su Copia nella cronologia corrente
Change Case con i seguenti parametri:
param-file“Da “: GSM461177_untreat_paired (output di Extract Datasettool)Gene length
“Change case of columns”: c1
“Delimited by”: Tab
“To”: Upper case
Rinominare l’output in Gene IDs and length
Ora abbiamo i due file di input necessari per goseq.
Pratica: Eseguire analisi GO
goseq ( Galaxy version 1.50.0+galaxy0) con
“Differentially expressed genes file”: Gene IDs and differential expression
“Gene lengths file”: Gene IDs and length
“Gene categories”: Get categories
“Select a genome to use”: Fruit fly (dm6)
“Select Gene ID format”: Ensembl Gene ID
“Select one or more categories”: GO: Cellular Component, GO: Biological Process, GO: Molecular Function
In “Output Options”
“Output Top GO terms plot?”: Yes
“Extract the DE genes for the categories (GO/KEGG terms)?”: Yes
goseq genera con questi parametri 3 output:
Una tabella (Ranked category list - Wallenius method) con le seguenti colonne per ogni termine GO:
category: Categoria GO
over_rep_pval: valore p per la sovrarappresentazione del termine nei geni differenzialmente espressi
under_rep_pval: valore p per la sottorappresentazione del termine nei geni differenzialmente espressi
numDEInCat: numero di geni differenzialmente espressi in questa categoria
numInCat: numero di geni in questa categoria
term: dettaglio del termine
ontology: MF (Molecular Function - attività molecolari dei prodotti genici), CC (Cellular Component - dove i prodotti genici sono attivi), BP (Biological Process - percorsi e processi più ampi costituiti dalle attività di più prodotti genici)
p.adjust.over_represented: valore p per la sovrarappresentazione del termine nei geni differenzialmente espressi, aggiustato per i test multipli con la procedura di Benjamini-Hochberg
p.adjust.under_represented: valore p per la sottorappresentazione del termine nei geni differenzialmente espressi, aggiustato per i test multipli con la procedura di Benjamini-Hochberg
Per identificare le categorie significativamente arricchite/non arricchite al di sotto di un certo cutoff p-value, è necessario utilizzare il valore p aggiustato.
Domanda
Quanti termini GO sono sovrarappresentati con un valore P aggiustato < 0,05? Quanti sono sottorappresentati?
Come vengono suddivisi i termini GO sovrarappresentati in MF, CC e BP? E per i termini GO sottorappresentati?
60 termini GO (0,50%) sono sovrarappresentati e 7 (0,07%) sottorappresentati.
Filtrare i dati su qualsiasi colonna usando semplici espressioni su c8 (valore p aggiustato per i termini GO sovrarappresentati) e c9 (valore p aggiustato per i termini GO sottorappresentati)
Per i sovrarappresentati, 50 BP, 5 CC e 5 MF e per i sottorappresentati, 5 BP, 2 CC e 0 MF
Grouping data sulla colonna 7 (categoria) e conteggio sulla colonna 1 (IDs)
Un grafico con i 10 termini GO più sovrarappresentati
Domanda
Cos’è l’asse x? Come viene calcolato?
L’asse delle ascisse è la percentuale di geni della categoria che sono stati identificati come differenzialmente espressi: \(100 \times \frac{numDEInCat}{numInCat}\)
Una tabella con i geni differenzialmente espressi (dall’elenco che abbiamo fornito) associati ai termini GO (DE genes for categories (GO/KEGG terms))
In questa esercitazione abbiamo trattato l’analisi dell’arricchimento di GO con goseq. Per conoscere altri metodi e strumenti per l’analisi dell’arricchimento dei gruppi di geni, consultare il tutorial “RNA-Seq genes to pathways”.
Analisi dei percorsi KEGG
goseq può essere utilizzato anche per identificare percorsi KEGG interessanti. Il database dei percorsi KEGG è una raccolta di mappe di percorsi che rappresentano le conoscenze attuali delle reti di interazione, reazione e relazione molecolare. Una mappa può integrare molte entità, tra cui geni, proteine, RNA, composti chimici, glicani e reazioni chimiche, nonché geni di malattie e bersagli di farmaci.
Per esempio, il percorso dme00010 rappresenta il processo di glicolisi (conversione del glucosio in piruvato con generazione di piccole quantità di ATP e NADH) per Drosophila melanogaster:
Pratica: Eseguire l'analisi dei percorsi KEGG
goseq ( Galaxy version 1.50.0+galaxy0) with
“Differentially expressed genes file”: Gene IDs and differential expression
“Gene lengths file”: Gene IDs and length
“Gene categories”: Get categories
“Select a genome to use”: Fruit fly (dm6)
“Select Gene ID format”: Ensembl Gene ID
“Select one or more categories”: KEGG
In “Output Options”
“Output Top GO terms plot?”: No
“Extract the DE genes for the categories (GO/KEGG terms)?”: Yes
goseq genera con questi parametri 2 output:
Una grande tabella con i termini KEGG e alcune statistiche
Domanda
Quanti termini dei percorsi KEGG sono stati identificati?
Quanti termini dei percorsi KEGG sono sovrarappresentati con un valore P aggiustato < 0,05?
Quali sono i termini dei percorsi KEGG sovrarappresentati?
Quanti termini dei percorsi KEGG sono sottorappresentati con un valore P aggiustato < 0,05?
Il file ha 128 righe, compresa l’intestazione, quindi sono stati identificati 127 percorsi KEGG.
2 percorsi KEGG (2,34%) sono sovrarappresentati, usando lo strumento Filtra i dati su qualsiasi colonna usando espressioni semplici su c6 (valore p aggiustato per i percorsi KEGG sovrarappresentati)
I 2 percorsi KEGG sovrarappresentati sono 01100 e 00010. Effettuando una ricerca su KEGG database, possiamo trovare ulteriori informazioni su questi percorsi:01100 corrisponde a tutte le vie metaboliche e00010 alla via della glicolisi/gluconeogenesi.
Nessun percorso KEGG è sottorappresentato, usando lo strumento Filtra i dati su qualsiasi colonna usando espressioni semplici su c7 (valore p aggiustato per i percorsi KEGG sottorappresentati)
Una tabella con i geni differenzialmente espressi (dall’elenco fornito) associati ai percorsi KEGG (DE genes for categories (GO/KEGG terms))
Si potrebbe indagare su quali geni sono coinvolti in quali percorsi esaminando il secondo file generato da goseq. Tuttavia, questa operazione può essere macchinosa e vorremmo vedere i percorsi come rappresentati nell’immagine precedente. Pathview (Luo and Brouwer 2013) può aiutare a generare automaticamente immagini simili alla precedente, aggiungendo anche informazioni aggiuntive sui geni (ad esempio l’espressione) nel nostro studio.
Questo strumento ha bisogno di 2 input principali:
ID del percorso da tracciare, o come un solo ID o come un file con una colonna con gli ID del percorso
Un file tabellare con i geni dell’esperimento RNA-Seq con 2 (o più) colonne:
gli ID dei geni (univoci all’interno del file)
alcune informazioni sui geni
Può trattarsi, ad esempio, di un valore di p o di un fold change. Queste informazioni saranno aggiunte al grafico del percorso: il nodo del gene corrispondente sarà colorato in base al valore. Se ci sono colonne diverse, le diverse informazioni verranno tracciate una accanto all’altra sul nodo.
Qui vorremmo visualizzare i 2 percorsi KEGG: il 00010 sovrarappresentato (Glicolisi / Gluconeogenesi) e il 03040 più sottorappresentato (ma non in modo significativo) (Spliceosoma). Vorremmo che i nodi dei geni fossero colorati in base al Log2 Fold Change per i geni differenzialmente espressi a causa del trattamento.
Pratica: Overlay log2FC su KEGG pathway
Cut colonne da una tabella con i seguenti parametri:
“Cut columns”: c1,c3
“Delimited by”: Tab
param-file“From”: Genes with significant adj p-value
Rinominare in Genes with significant adj p-value and their Log2 FC
Abbiamo estratto l’ID e il Log2 Fold Change per i geni che hanno un valore p aggiustato significativo.
Creare un nuovo file di tabelle con i seguenti dati (ID dei percorsi da tracciare) denominati KEGG pathways to plot
00010
03040
Pathview ( Galaxy version 1.34.0+galaxy0) with
“Number of pathways to plot”: Multiple
param-file“KEGG pathways”: KEGG pathways to plot
“Does the file have header (a first line with column names)?”: No
“Species to use”: Fly
“Provide a gene data file?”: Yes
param-file“Gene data”: Genes with significant adj p-value and their Log2 FC
“Does the file have header (a first line with column names)?”: Yes
“Format for gene data”: Ensembl Gene ID
“Provide a compound data file?”: No
In “Output Options”
“Output for pathway”: KEGG native
“Plot on same layer?”: Yes
Pathview genera una raccolta con la visualizzazione KEGG: un file per percorso.
Domanda
dme00010 percorso KEGG da Pathview
Cosa sono le caselle colorate?
Qual è il codice colore?
Le caselle colorate sono i geni del percorso espressi in modo differenziato
Prestare attenzione al fatto che il codice dei colori è controintuitivo: il verde è per i valori inferiori a 0, significa per i geni con un log2FC < 0 e il rosso per i geni con un log2FC > 0.
Conclusione
In questo tutorial, abbiamo analizzato dati reali di sequenziamento dell’RNA per estrarre informazioni utili, come ad esempio quali geni sono aumentati o diminuiti dalla deplezione del gene Pasilla, ma anche in quali termini GO o percorsi KEGG sono coinvolti. Per rispondere a queste domande, abbiamo analizzato i set di dati di sequenze di RNA utilizzando un approccio di analisi dei dati RNA-Seq basato su riferimenti. Questo approccio può essere riassunto con il seguente schema:
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Levin, J. Z., M. Yassour, X. Adiconis, C. Nusbaum, D. A. Thompson et al., 2010 Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods 7: 709. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3005310/
Brooks, A. N., L. Yang, M. O. Duff, K. D. Hansen, J. W. Park et al., 2011 Conservation of an RNA regulatory map between Drosophila and mammals. Genome Research 21: 193–202. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3032923/
Kim, D., B. Langmead, and S. L. Salzberg, 2015 HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12: 357. https://www.nature.com/articles/nmeth.3317
Ewels, P., M. Magnusson, S. Lundin, and M. K\~ A\textcurrencyller, 2016 MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048. 10.1093/bioinformatics/btw354
Kim, D., J. M. Paggi, C. Park, C. Bennett, and S. L. Salzberg, 2019 Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37: 907–915. https://www.nature.com/articles/s41587-019-0201-4
Feedback
Hai usato questo materiale come istruttore? Sentiti libero di lasciarci un feedback. Com'è andata.
Hai usato questo materiale come studente? Clicca sul modulo qui sotto per lasciare un feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{transcriptomics-ref-based,
author = "Bérénice Batut and Mallory Freeberg and Mo Heydarian and Anika Erxleben and Pavankumar Videm and Clemens Blank and Maria Doyle and Nicola Soranzo and Peter van Heusden and Lucille Delisle",
title = "Analisi dei dati RNA-Seq basata su riferimenti (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/ref-based/tutorial_IT.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Riferimenti
Queste persone o organizzazioni hanno fornito supporto finanziario per lo sviluppo di questa risorsa
Domande:

Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab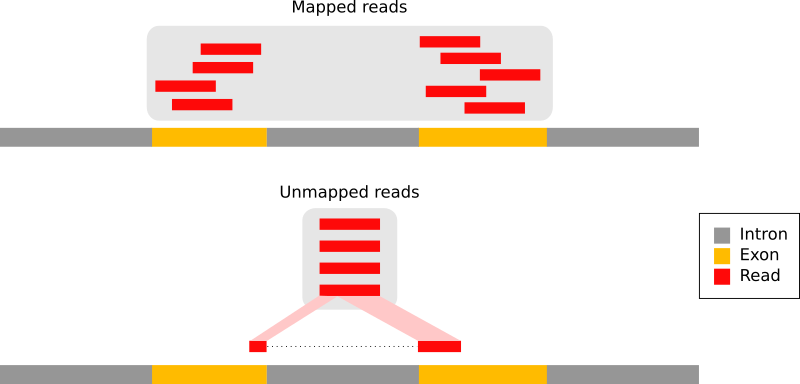 Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab

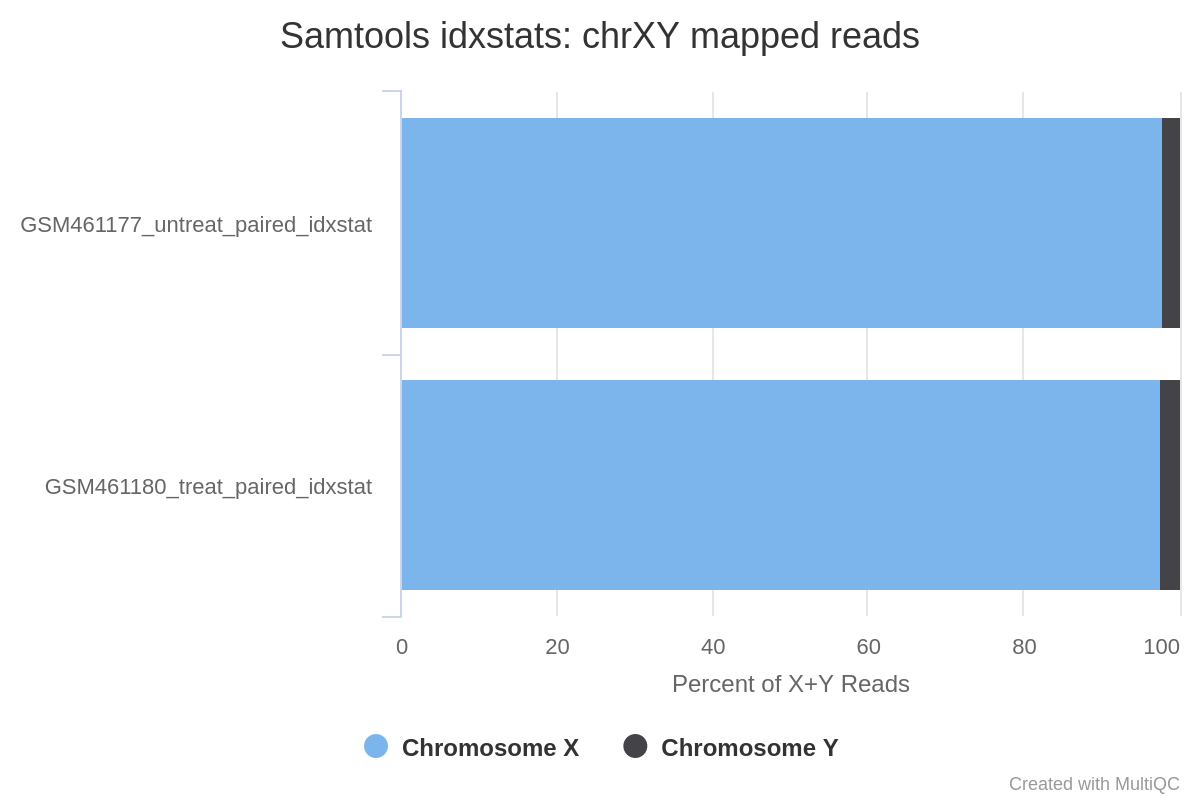
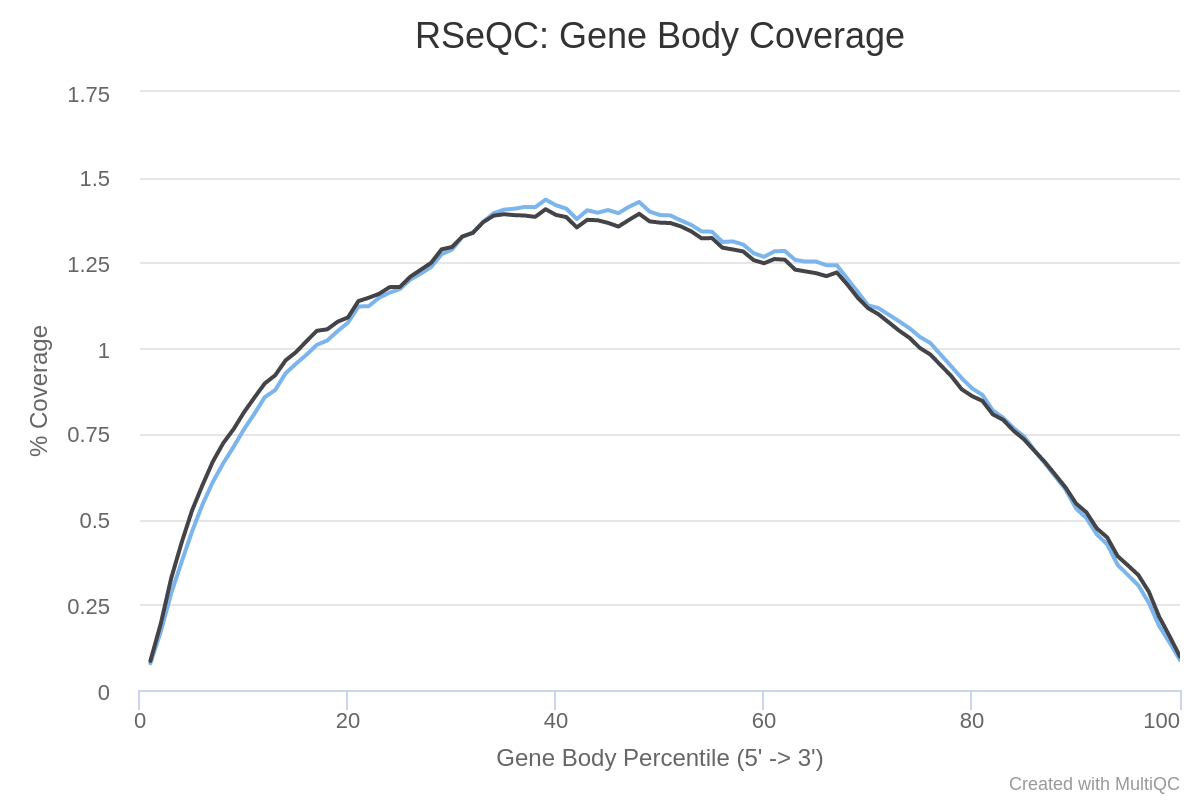
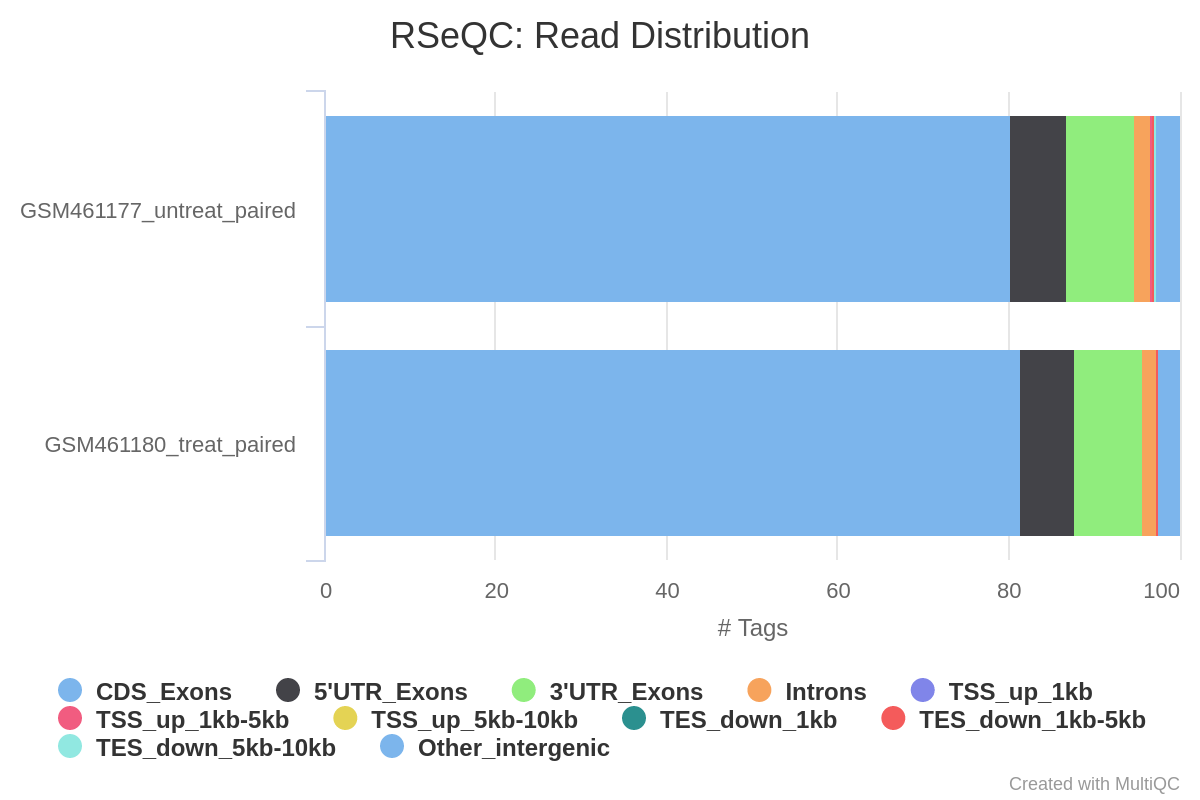
 Open image in new tab
Open image in new tab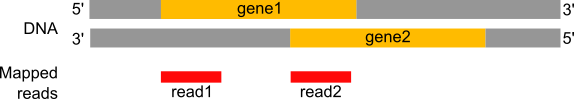 Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
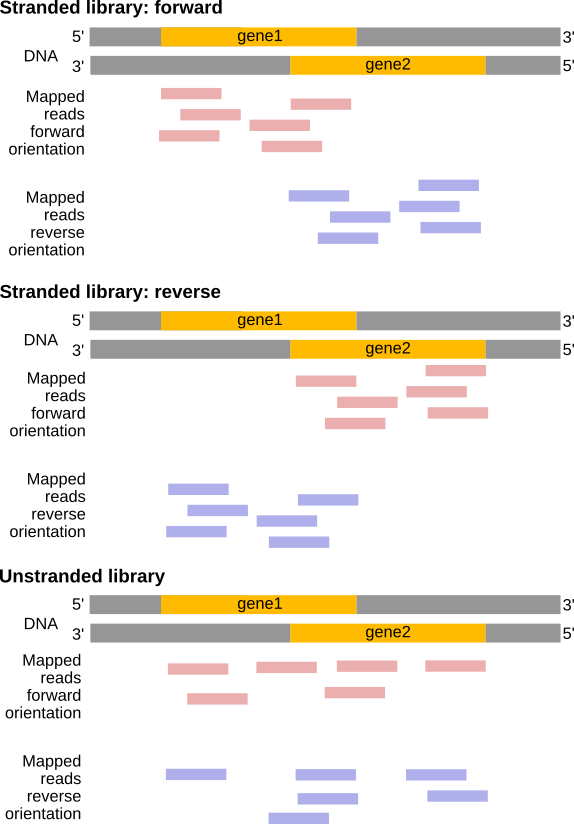 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab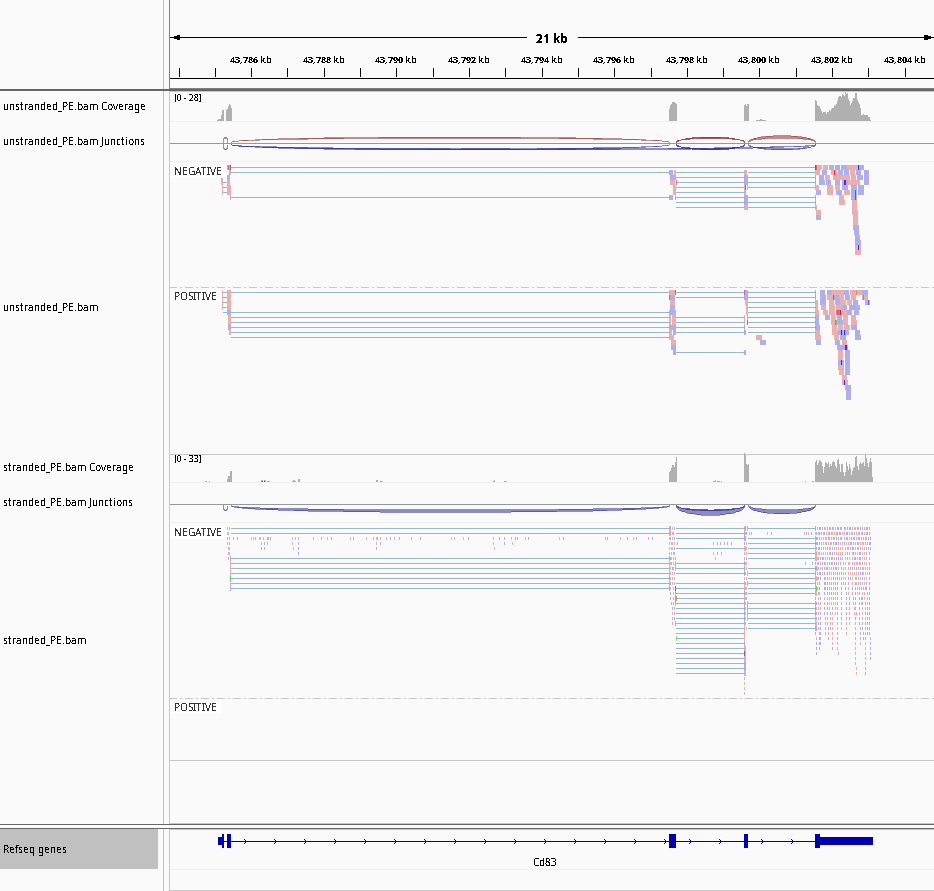 Open image in new tab
Open image in new tab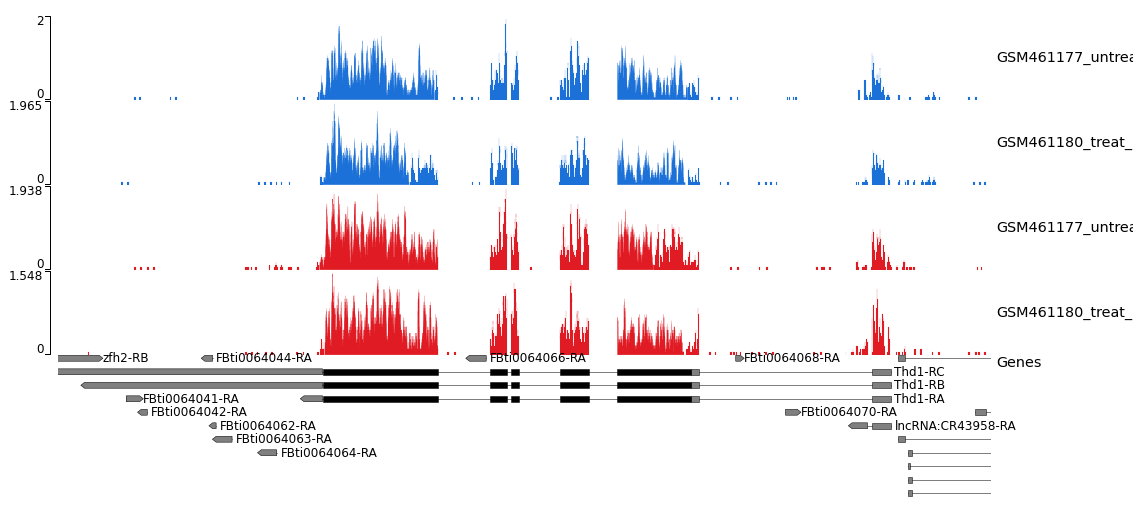 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab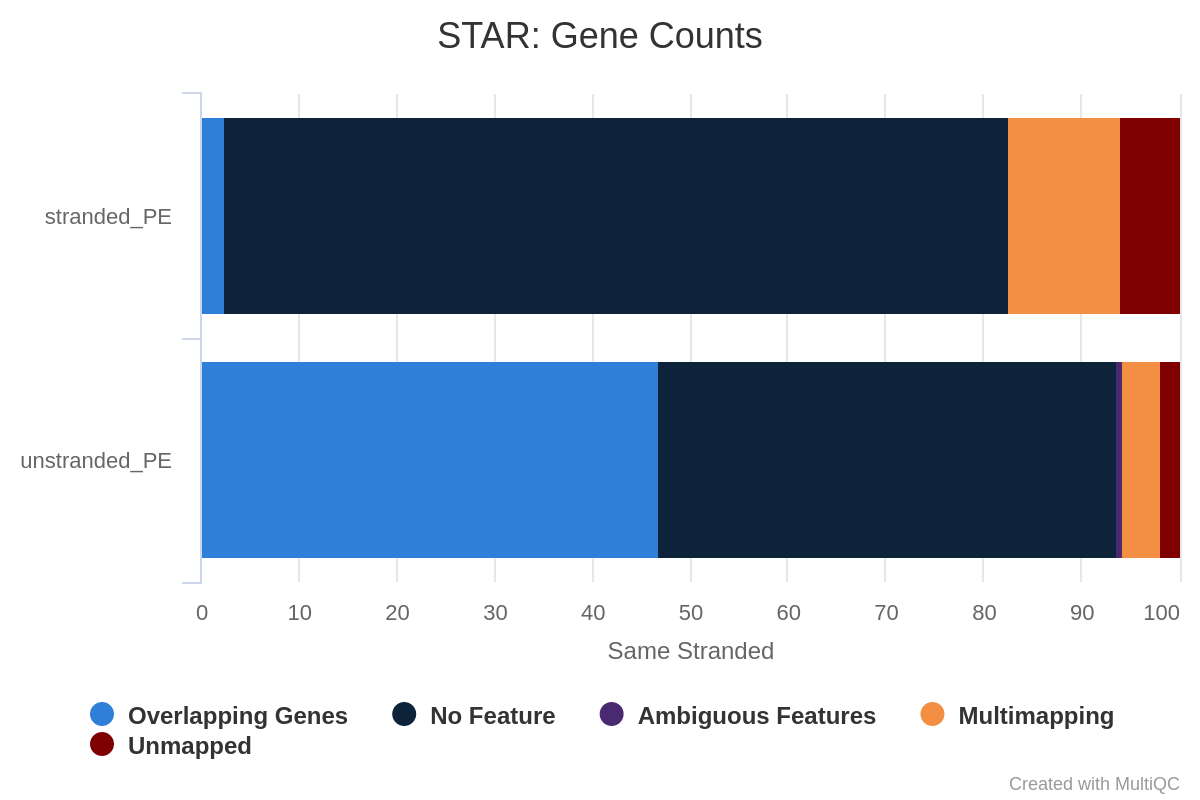 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab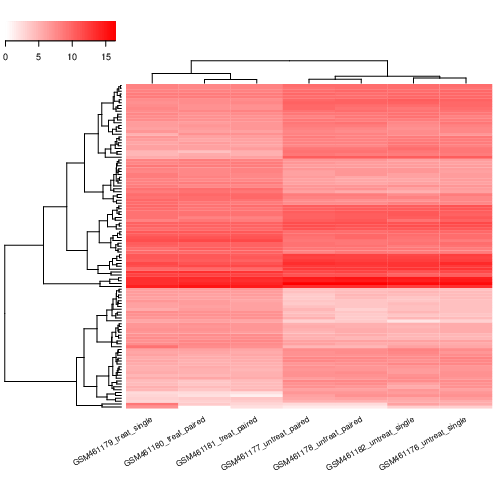 Open image in new tab
Open image in new tab Open image in new tab
Open image in new tab


 Open image in new tab
Open image in new tab
